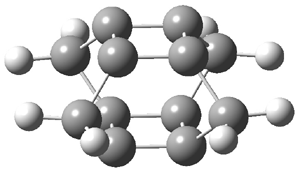
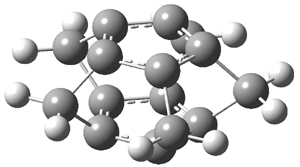
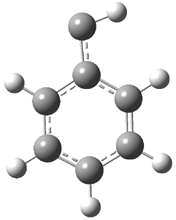
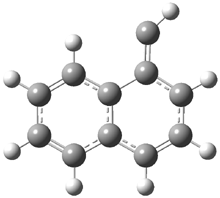
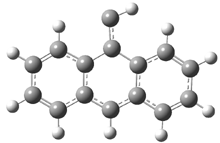
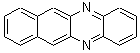
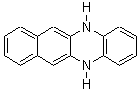
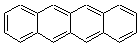
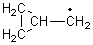Schleyer continues his study of aromaticity with a paper1 that picks up on the theme presented in one2 I have previously blogged on – the relationship between a formally aromatic pyrazine and formally antiaromatic dihydropyrazine. He now examines the diazotetracene 1 and it dihydro analogue 2.1 In terms of formal electron count, 1 should be aromatic, just like the all carbon analogue tetracene 3, and 2 should be antiaromatic.
Schleyer used the NICSπzz values obtained in the center of each ring to evaluate the aromatic/antiaromatic character of these three molecules. These calculations were performed using canonical molecular orbitals and repeated using localized molecular orbitals. The results are similar for each method, and the canonical MO values are presented in Table 1. As expected for an aromatic compound, each ring of tetracene 3 has large negative NICS values, indicating that each ring is locally aromatic and the molecule as a whole is aromatic. The same is true for the diazotetracene 1. (In fact the NICS values for 1 and 3 are remarkably similar.) However, for 2, the dihydropyrazine ring has a positive NICS values, indicative of a locally antiaromatic ring. While the three phenyl rings have negative NICS values, these absolute values are smaller than for the rings of 1 or 3, indicating an attenuation of their aromaticity. Nonetheless, the sum of the NICS values of 2 is negative, suggesting that the molecule is globally aromatic, though only marginally so. This is due to the antiaromaticity of the dihydropyrazine ring being delocalized to some extent over the entire molecule. Schleyer, concludes that “large 4n π compounds […] are not appreciably destabilized relative to their 4n+2 π congeners.”
Table 1 NICSπzz (ppm) for each ring of 1-3 and their sum.1
|
|
||||
|
-30.0 |
-42.5 |
-41.1 |
-30.1 |
sum = -144.0 |
|
|
||||
|
-26.3 |
-14.2 |
31.3 |
-16.7 |
sum = -25.9 |
|
|
||||
|
-29.6 |
-42.1 |
-42.1 |
-29.6 |
Sum = -143.4 |
References
(1) Miao, S.; Brombosz, S. M.; Schleyer, P. v. R.; Wu, J. I.; Barlow, S.; Marder, S. R.; Hardcastle, K. I.; Bunz, U. H. F., "Are N,N-Dihydrodiazatetracene Derivatives Antiaromatic?," J. Am. Chem. Soc., 2008, 130, 7339-7344, DOI: 10.1021/ja077614p.
(2) Miao, S.; Schleyer, P. v. R.; Wu, J. I.; Hardcastle, K. I.; Bunz, U. H. F., "A Thiadiazole-Fused N,N-Dihydroquinoxaline: Antiaromatic but Isolable," Org. Lett. 2007, 9, 1073-1076, DOI: 10.1021/ol070013i
InChIs
1: InChI=1/C18H12/c1-2-6-14-10-18-12-16-8-4-3-7-15(16)11-17(18)9-13(14)5-1/h1-12H
2: InChI=1/C16H10N2/c1-2-6-12-10-16-15(9-11(12)5-1)17-13-7-3-4-8-14(13)18-16/h1-10H
3: InChI=1/C16H12N2/c1-2-6-12-10-16-15(9-11(12)5-1)17-13-7-3-4-8-14(13)18-16/h1-10,17-18H