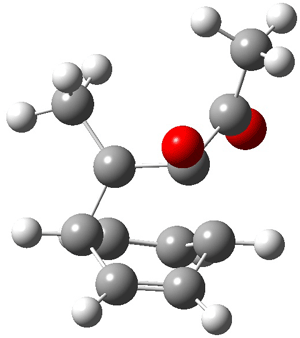
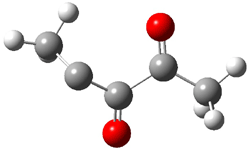
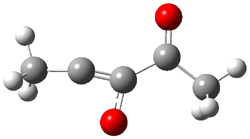
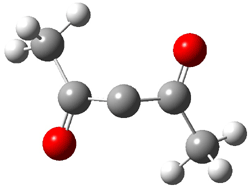
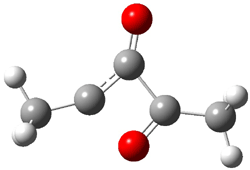
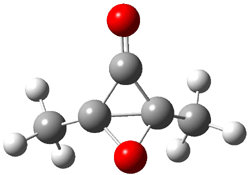
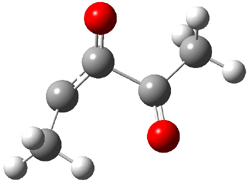
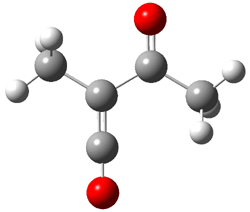
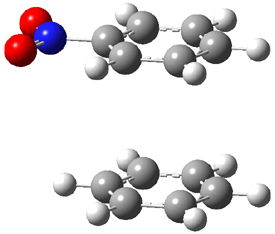
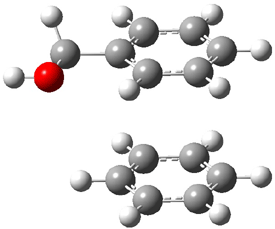
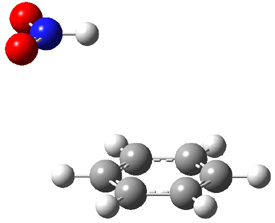
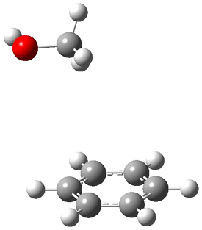
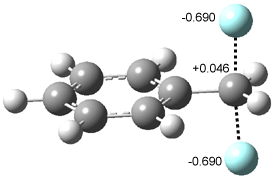
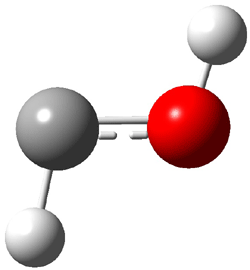
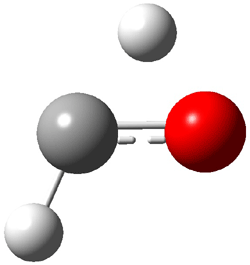
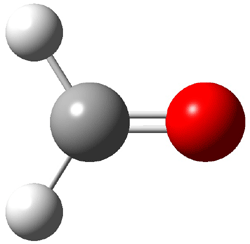
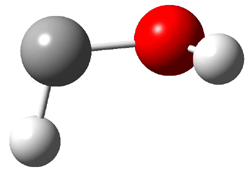
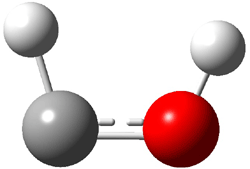
An alternative take on the nature of the interaction in π-stacking is offered by Wheeler and Houk.1 They start by examining the binding between benzene and a series of 24 substituted benzenes. Two representative dimmers are shown in Figure 1, where the substituent is NO2 or CH2OH. As was noted in a number of previous studies,2-6 the binding with any substituted benzene is stronger than the parent benzene dimer. Nonetheless, Wheeler and Houk point out that the binding energy has a reasonable correlation with σm. It appears that the benzene dimer itself is the outlier; the binding energy when the substituent is CH2OH, whose σm value is zero, is bound more tightly than the benzene dimer. They conclude that there is a dispersive interaction between any substituent and the other benzene ring.
Figure 1. MO5-2X/6-31+G(d) optimized geometries of (a) C6H6-C6H5NO2, (b) C6H6-C6H5CH2OH, (c) C6H6-HNO2, and (d) C6H6-HCH2OH.1
They next constructed an admittedly very crude model system whereby the substituted benzene C6H5X is replaced by HX; the corresponding models are also shown in Figure 1. The binding energies of these model dimmers correlates very well with the real dimmers, with r = 0.91. Rather than involving the interaction of the π-electrons, the origin of the enhanced binding in aromatic dimers involves electrostatic interactions of the substituent with the other aromatic ring – effectively the quadrupole of the unsubstituted ring interacts with the dipoles of the substituent and its ring system. In addition, the inherent dispersive interaction increase the binding.
References
(1) Wheeler, S. E.; Houk, K. N., "Substituent Effects in the Benzene Dimer are Due to Direct Interactions of the Substituents with the Unsubstituted Benzene," J. Am. Chem. Soc., 2008, 130, 10854-10855, DOI: 10.1021/ja802849j.
(2) Sinnokrot, M. O.; Sherrill, C. D., "Unexpected Substituent Effects in Face-to-Face π-Stacking Interactions," J. Phys. Chem. A, 2003, 107, 8377-8379, DOI: 10.1021/jp030880e.
(3) Sinnokrot, M. O.; Sherrill, C. D., "Substituent Effects in π-&pi Interactions: Sandwich and T-Shaped Configurations," J. Am. Chem. Soc., 2004, 126, 7690-7697, DOI: 10.1021/ja049434a.
(4) Sinnokrot, M. O.; Sherrill, C. D., "Highly Accurate Coupled Cluster Potential Energy Curves for the Benzene Dimer: Sandwich, T-Shaped, and Parallel-Displaced Configurations," J. Phys. Chem. A, 2004, 108, 10200-10207, DOI: 10.1021/jp0469517
(5) Lee, E. C.; Kim, D.; Jurecka, P.; Tarakeshwar, P.; Hobza, P.; Kim, K. S., "Understanding of Assembly Phenomena by Aromatic-Aromatic Interactions: Benzene Dimer and the Substituted Systems," J. Phys. Chem. A 2007, 111, 3446-3457, DOI: 10.1021/jp068635t.
(6) Grimme, S.; Antony, J.; Schwabe, T.; Mück-Lichtenfeld, C., "Density functional theory with dispersion corrections for supramolecular structures, aggregates, and complexes of
(bio)organic molecules," Org. Biomol. Chem. 2007, 741-758, DOI: 10.1039/b615319b