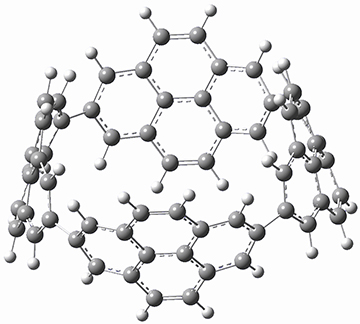
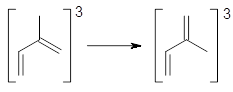
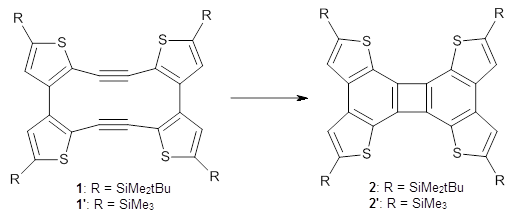
Aromaticity and orbital symmetry rules, though seemingly of ancient origin, remain areas of active interest. This paper by Fukazawa, et al combine both issues.1 The multiple-step electrocyclization of 1 gives 2 in a reaction that takes 9 days at 80 °C. What would be the effect of diminishing the aromatic character of the fused rings of 1? Would the reaction be faster or slower?
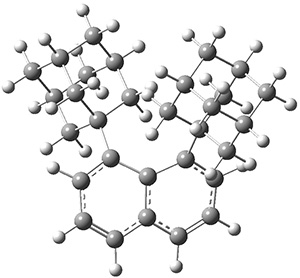
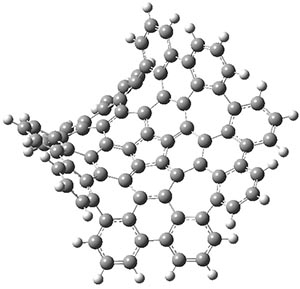
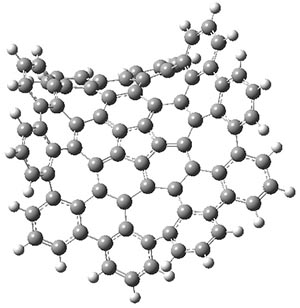
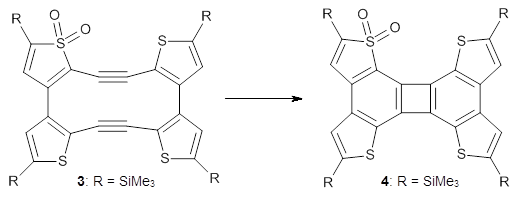
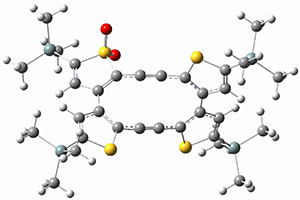
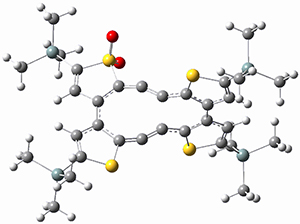
Before discussing the experimental results, let’s examine the B3LYP/6-31G(d) results for the reaction of 1’, 3 and 5. (Note that a slightly smaller pendant substituent is used in the computations than in the experiment.) The optimized geometries of the critical points along the reaction pathway for the cyclization of 3 are shown in Figure 1.
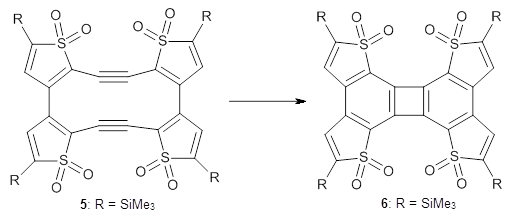
3 |
3-TS1 |
3-INT |
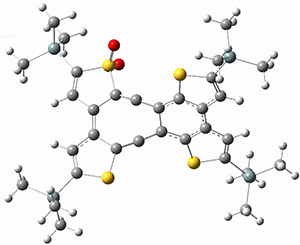
3-TS2 |
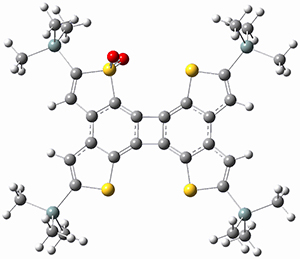
4 |
|
Figure 1. B3LYP/6-31G(d) optimized geometries and relative energies (kcal mol-1) for the critical points along the reaction 3 → 4.
Remember that all structures on my blog can be viewed interactively by clicking on the image of the molecule.
For 1’, the first barrier (for the 8π cyclization) has a barrier of about 23 kcal mol-1, but the second step (the 4π cyclization) has an even larger barrier of 28 kcal mol-1. However, reducing the aromaticity of one of the fused rings (compound 3) leads to lower barriers of 18 and 13 kcal mol-1. For the cyclization of 5, only a single transition state was found – no intermediate and no second TS – with a barrier of 12 kcal mol-1. Thus, removing these external aromatic rings reduces the barrier of the reaction, and that is exactly what is found experimentally!
References
(1) Fukazawa, A.; Oshima, H.; Shimizu, S.; Kobayashi, N.; Yamaguchi, S. "Dearomatization-Induced Transannular Cyclization: Synthesis of Electron-Accepting Thiophene-S,S-Dioxide-Fused Biphenylene," J. Am. Chem. Soc. 2014, 136, 8738-8745, DOI: 10.1021/ja503499n.
InChIs:
1: InChI=1S/C44H64S4Si4/c1-41(2,3)49(13,14)37-25-29-30-26-38(50(15,16)42(4,5)6)46-34(30)23-24-36-32(28-40(48-36)52(19,20)44(10,11)12)31-27-39(51(17,18)43(7,8)9)47-35(31)22-21-33(29)45-37/h25-28H,1-20H3/b30-29-,32-31-
InChIKey=OCNQBMWQONUVNH-IOYDOZLVSA-N
1’:InChI=1S/C32H40S4Si4/c1-37(2,3)29-17-21-22-18-30(38(4,5)6)34-26(22)15-16-28-24(20-32(36-28)40(10,11)12)23-19-31(39(7,8)9)35-27(23)14-13-25(21)33-29/h17-20H,1-12H3/b22-21-,24-23-
InChIKey=GTFPBRMBCLREPG-ICHHBZPXSA-N
2: InChI=1S/C44H64S4Si4/c1-41(2,3)49(13,14)29-21-25-26-22-30(50(15,16)42(4,5)6)46-38(26)34-33(37(25)45-29)35-36(34)40-28(24-32(48-40)52(19,20)44(10,11)12)27-23-31(47-39(27)35)51(17,18)43(7,8)9/h21-24H,1-20H3
InChIKey=OTDXAOVIIQYYNV-UHFFFAOYSA-N
2’: InChI=1S/C32H40S4Si4/c1-37(2,3)21-13-17-18-14-22(38(4,5)6)34-30(18)26-25(29(17)33-21)27-28(26)32-20(16-24(36-32)40(10,11)12)19-15-23(35-31(19)27)39(7,8)9/h13-16H,1-12H3
InChIKey=IYZNCPPDTHWWCO-UHFFFAOYSA-N
3: InChI=1S/C32H40O2S4Si4/c1-39(2,3)29-17-21-22-18-30(40(4,5)6)37-27(22)15-16-28-24(20-32(38(28,33)34)42(10,11)12)23-19-31(41(7,8)9)36-26(23)14-13-25(21)35-29/h17-20H,1-12H3/b22-21-,24-23-
InChIKey=ZJBDGDJVLGNVOD-ICHHBZPXSA-N
4: InChI=1S/C32H40O2S4Si4/c1-39(2,3)21-13-17-18-14-22(40(4,5)6)36-30(18)26-25(29(17)35-21)27-28(26)32-20(16-24(38(32,33)34)42(10,11)12)19-15-23(37-31(19)27)41(7,8)9/h13-16H,1-12H3
InChIKey=QUSJUOMZBJUGON-UHFFFAOYSA-N
5: InChI=1S/C32H40O8S4Si4/c1-45(2,3)29-17-21-22-18-30(46(4,5)6)42(35,36)26(22)15-16-28-24(20-32(44(28,39)40)48(10,11)12)23-19-31(47(7,8)9)43(37,38)27(23)14-13-25(21)41(29,33)34/h17-20H,1-12H3/b22-21-,24-23-
InChIKey=NNZTUSIYEPMHMP-ICHHBZPXSA-N
6: InChI=1S/C32H40O8S4Si4/c1-45(2,3)21-13-17-18-14-22(46(4,5)6)42(35,36)30(18)26-25(29(17)41(21,33)34)27-28(26)32-20(16-24(44(32,39)40)48(10,11)12)19-15-23(47(7,8)9)43(37,38)31(19)27/h13-16H,1-12H3
InChIKey=JZHQQYXUIQXWLQ-UHFFFAOYSA-N