Naphthalene, phenanthrene and pyrene are all planar aromatic compounds. How can substituted version be chiral, with the chirality present in the aromatic portion of the molecule? The answer is provided by Yamaguchi and Kwon.1 They prepared peri-substituted analogues with the bulky adamantly group as the substituents. This bulky requires one adamantyl group to be position above the aromatic plane and the other below the plane, as in 1 and 2.
|
|
|
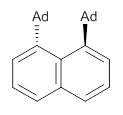
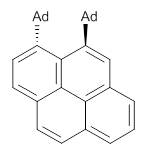
These molecules and two other examples were prepared in their optically pure form. B3LYP/6-31G(d) computations were performed on both of these structures (shown in Figure 1), but computations are a minor component of the work. These structures do show the out-of-plane distortions at C1 and C8, also apparent in the crystal structures. Computations of naphthalene and 1,8-dimethylnaphthalene show a planar naphthalene backbone, but i-propyl substitution does force the substituents out of plane.
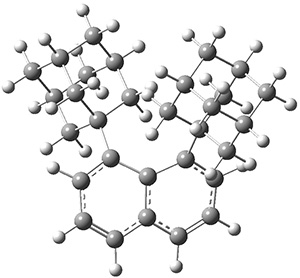 1 |
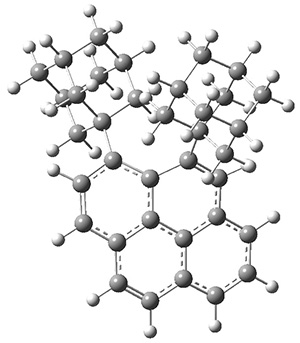 2 |
Figure 1. B3LYP/6-31G(d) optimized structures of 1 and 2.
These types of systems continue to subject the notion of “aromaticity” to serious scrutiny.
References
(1) Yamamoto, K.; Oyamada, N.; Xia, S.; Kobayashi, Y.; Yamaguchi, M.; Maeda, H.; Nishihara, H.; Uchimaru, T.; Kwon, E. "Equatorenes: Synthesis and Properties of Chiral Naphthalene, Phenanthrene, Chrysene, and Pyrene Possessing Bis(1-adamantyl) Groups at the Peri-position," J. Am. Chem. Soc. 2013, 135, 16526-16532, DOI: 10.1021/ja407800e.
InChIs
1: InChI=1S/C30H36/c1-3-25-4-2-6-27(30-16-22-10-23(17-30)12-24(11-22)18-30)28(25)26(5-1)29-13-19-7-20(14-29)9-21(8-19)15-29/h1-6,19-24H,7-18H2
InChIKey=QNPJKZPPLCPHSS-UHFFFAOYSA-N
2: InChI=1S/C36H38/c1-2-27-4-5-28-6-7-30(35-15-21-8-22(16-35)10-23(9-21)17-35)34-31(14-29(3-1)32(27)33(28)34)36-18-24-11-25(19-36)13-26(12-24)20-36/h1-7,14,21-26H,8-13,15-20H2
InChIKey=DKRBDGNWYTWNHL-UHFFFAOYSA-N

Henry Rzepa responded on 11 Jan 2014 at 11:21 am #
Chiral aromaticity is not entirely new! There are a whole series of 4n+2 aromatics that are chiral. The trick of course is that the linking number Lk for the π system topology is ≠ 0. Mostly commonly for the chiral aromatics, Lk =± 2π (this number is also a chiral descriptor, in the Cahn-Ingold-Prelog sense, the two enantiomers having opposite linking numbers). See for example: doi: 10.1021/ol901172g (2009).
In that sense, the benzenoid aromatics are the exception rather than the rule!
Alireza Marefat khah responded on 12 Jan 2014 at 11:16 am #
There is an extra Hydrogen, a -C-H-H connection, in the structure number one (should see it if turn the #1 a bit). Where does it come from? Is it just a typo in the blog or the structures were transferred from the supplementary materials of the original article?
Best regards,
Alireza
Steven Bachrach responded on 13 Jan 2014 at 8:46 am #
There actually isn’t an extra hydrogen, it’s just in the wrong place. This happened in getting the coordinates from the supporting materials – copying and pasting had every negative sign from the supporting materials converted over into a “0” when pasted into the editor. This sort of thing happens so frequently!
Anyways, it is fixed now – you may have to clean your cache to make sure you’re picking up a fresh copy of the coordinated.
Thanks for bringing this to my attention!
Henry Rzepa responded on 16 Jan 2014 at 4:44 am #
Steve,
There is some confusion regarding the use of Java/Jmol now. A spate of recent security releases for the run-time environment on desktop computers has meant that only digitally signed applets will be problem free. Yesterday for example, Java 7, update 51 landed on many computers. I found that your Jmol (version 11.6.21 dating from 2008) ran, albeit with fairly intimidating warnings. It might be that in the near future it will not run at all (and this might be OS dependent as well).
Jmol 14.0.5 now comes as a suitably signed applet, and the user need only accept it once for each site which invokes it.
Some erstwhile Java-based sites have now migrated to JSmol, which does not depend on Java. That solves the issues of course, but it (currently) brings with it a new problem, which is that JSmol is slower than Jmol (for some types of rendering, up to 25 times slower). JSmol has another advantage, which is that its access to the local file system of a computer is less restricted than the model imposed by Java.
As I noted, my Mac using Safari and Java 7, update 51 did manage to load your Jmol. It might be worth asking what other people’s experience has been?
In the longer (medium? Short?) term, some action is probably needed by any site that continues to run anything less than the very latest (14.0.5) version of Jmol.
Henry Rzepa responded on 24 Jan 2014 at 1:36 am #
Steve,
This comment was posted on my blog. It represents a way of globally editing the MySQL database off which blogs like this one operate to e.g. update how Jmol might be invoked.
You might also be interested in some simple benchmarks I am accumulating for the performance of eg JSmol vs Jmol. The former, as an interpreted language, is always going to be slower than the compiled Java-based Jmol. For most simple molecule renderings, the slow down is actually inconsequential. For some operations however, such as surface rendering, it can become significant (I have seen 25-fold slowdowns). But one avoids very high resolution surfaces, and keeps to relatively low-resolution versions, even here JSmol is acceptably fast. And to be fair, the benchmarks seem to be halving (getting faster) at rather better than Moore’s Law! The 2013 versions of both browsers and hardware (tablets) are about twice as fast as the 2012 vintage, and no doubt the situation will continue into 2014 and beyond.