The concept of antiaromaticity is an outgrowth of the well-entrenched notion or aromaticity. While 4n+2 π-electron systems are aromatic, 4n π-electron systems should be antiaromatic. That should mean that antiaromatic systems are unstable. The cyclopropenyl anion 1a has 4 π-electrons and should be antiaromatic. Kass has provided computational results that strongly indicate it is not antiaromatic!1
Let’s first look at the 3-cyclopropenyl cation 1c. Kass has computed (at both G3 and W1) the hydride affinity of 1c-4c. The hydride affinities of the latter three compounds plotted against the C=C-C+ angle is linear. The hydride affinity of 1c however falls way below the line, indicative of 1c being very stable – it is aromatic having just 2 π-electrons.
A similar plot of the deprotonation enthalpies leading to 1a-4d vs. C=C-C– angle is linear including all four compounds. If 1a where antiaromatic, one would anticipate that the deprotonation energy to form 1a would be much greater than expected simply from the effect of the smaller angle. Kass suggests that this indicates that 1a is not antiaromatic, but just a regular run-of-the-mill (very) reactive anion.
A hint at what’s going on is provided by the geometry of the lowest energy structure of 1a, shown in Figure 1. The molecule is non-planar, having Cs symmetry. A truly antiaromatic structure should be planar, really of D3h symmetry. The distortion from this symmetry reduces the antiaromatic character, in the same way that cyclobutadiene is not a perfect square and that cyclooctatraene is tub-shaped and not planar. So perhaps it is more fair to say that 1a has a distorted structure to avoid antiaromaticity, and that the idealized D3h structure, does not exist because of its antiaromatic character.
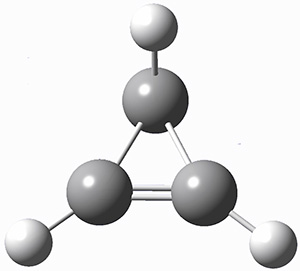
Figure 1. G3 optimized geometry of 1a.
References
(1) Kass, S. R. "Cyclopropenyl Anion: An Energetically Nonaromatic Ion," J. Org. Chem. 2013, 78, 7370-7372, DOI: 10.1021/jo401350m.
InChIs
1a: InChI=1S/C3H3/c1-2-3-1/h1-3H/q-1
InChIKey=IBTMQWIWZUYLHW-UHFFFAOYSA-N
1c: InChI=1S/C3H3/c1-2-3-1/h1-3H/q+1
InChIKey=IPKCFGQXHZKYLH-UHFFFAOYSA-N

Henry Rzepa responded on 24 Jan 2014 at 1:28 am #
You would find pretty much the same story told about avoided antiaromaticity in this anion in this blog post dated December 2010. I would add that one can regard this distortion as arising from a Jahn-Teller effect. In effect two electrons occupy a degenerate orbital pair (in D3h symmetry). For the closed shell singlet state, this implies that there are actually TWO ways of distorting, depending on which of the degenerate pair you put the two electrons into. Figure 1 above represents only one of the possible distortions. In the blog noted above you will find the other (and you can find more details of this form at this digital repository doi: 10042/to-6077). According to the calculations reported there, these two geometrical isomers are almost iso-energetic.
I would add that even the veritable cyclobutadiene (isoelectronic of course with the cyclopropene anion) has more ways of distorting and avoiding anti-aromaticity than meets the eye. Most would claim that there are just two rectangular forms which achieve this. In fact there is a third avoided form (Sept 2011). It can be seen by following this link and at this doi: 10.1002/chem.201102942. This form can be stabilised by setting up hydrogen bonding to do so (i.e. using the guanidinium cation to promote the effect, see digital repository entry 10042/to-9387). It is a bit higher in energy than the rectangular form, but it is still a clear cut geometrical isomer, which to my knowledge is neither widely known nor often considered.
Very few 4nπ-electron antiaromatics do not find some way of avoiding the anti-aromaticity. Geometric distortions, such as with the cyclopropenyl anion, are the most common. Extreme bond alternation is another method (although that in itself is not sufficient to attenuate the paratropicity). My own favourite type of molecule which cannot avoid anti-aromaticity is the lemniscular octaphyrin motif, an extended porphyrin which adopts the ∞ or lemniscular figure-eight shape. The very rigidity of this structural motif means that geometrical distortions to avoid anti-aromaticity in a 4n π-electron version cannot be achieved (other than by bond alternation). See doi: 10.1021/jo801022b.
Steven Bachrach responded on 24 Jan 2014 at 9:19 am #
I agree with you Henry in that your blog post really lays out the solution to this problem – the cyclopropenyl anion is not antiaromatic because it distorts its geometry to avoid being antiaromatic. The interesting contribution here by Steve Kass is the clever energetic analysis.