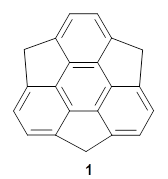
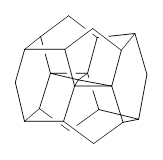
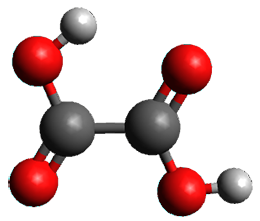
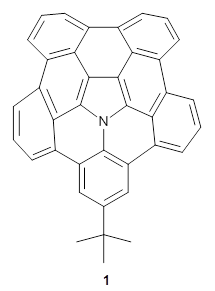
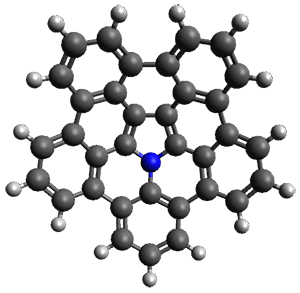
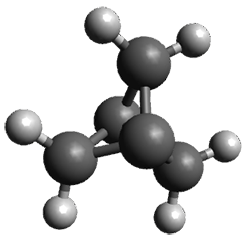
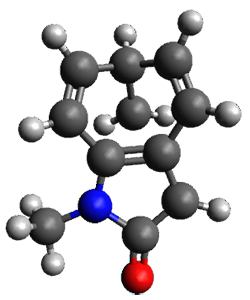
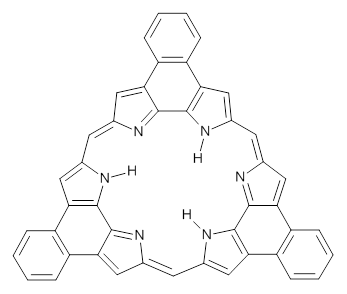
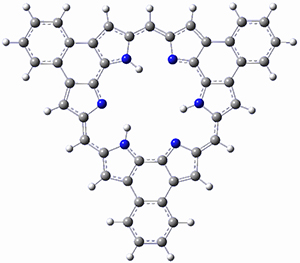
What is the spin state of the ground state of an aromatic species? Can this be spin state be manipulated by charge? These questions are addressed by Borden, Kim, Sessler and coworkers1 for the hexaphyrin 1. B3LYP/6-31G(d) optimization of 1 shows it to be a ground state singlet. This structure is shown in Figure 1.
|
|
|
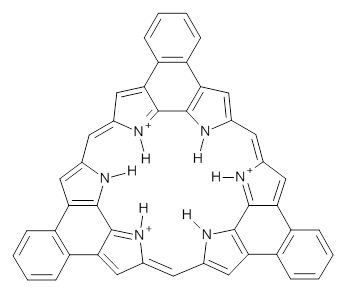
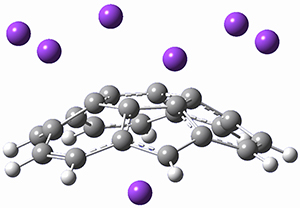
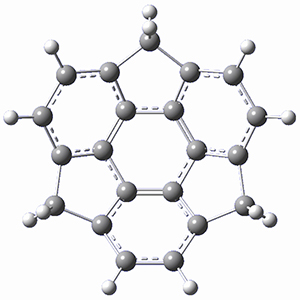
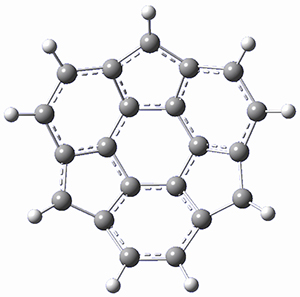
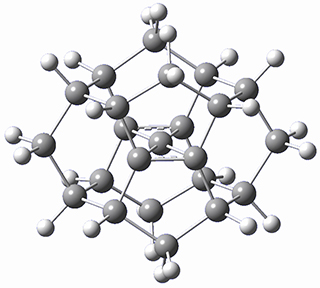
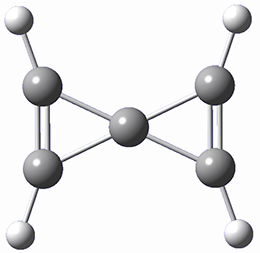
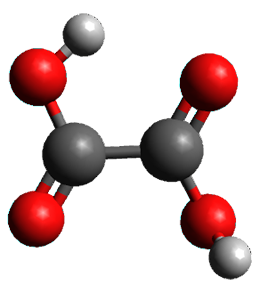
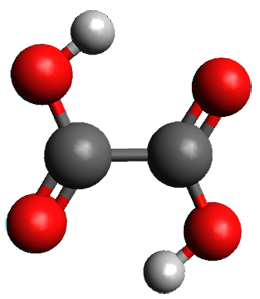
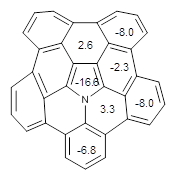
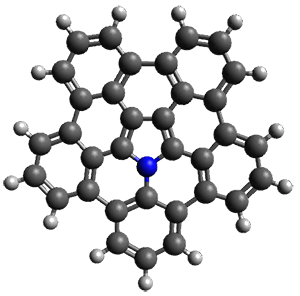
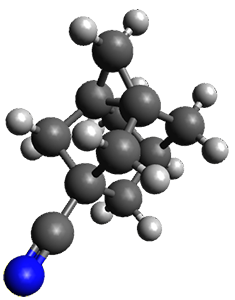
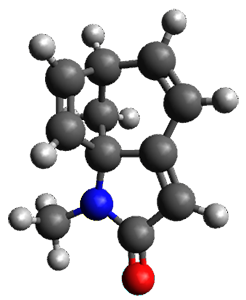
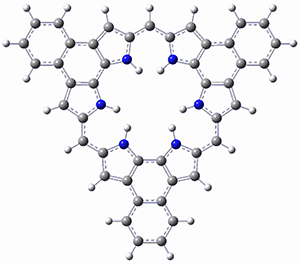
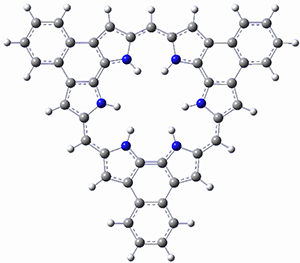
Protonation of the three pyrrole nitrogens creates 13+, which has interesting frontier orbitals. The HOMO of 13+, of a1” symmetry, has nodes running through all six nitrogens. The next higher energy orbital, of a2” symmetry, has a small π-contribution on each nitrogen. Protonation will therefore have no effect on the energy of the a1” orbital, but the charge will stabilize the a2” orbital. This will lower the energy gap between the two orbitals, suggesting that a ground state triplet might be possible. The lowest singlet and triplet states of 13+ are also shown in Figure 1.
1 |
|
Singlet 13+ |
Triplet 13+ |
Figure 1. (U)B3LYP/6-31G(d) optimized structures of 1 and singlet and triplet 13+.
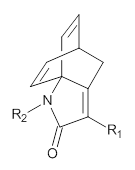
This spin state change upon protonation was experimentally verified by synthesis of two analogues of 1, shown below. The triprotonated versions of both are observed to have triplet character in their EPR spectrum.
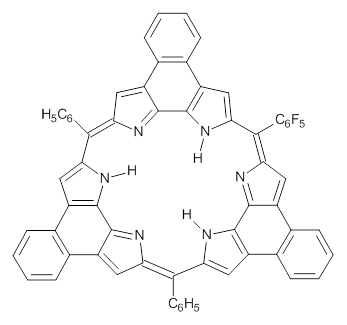
|
|
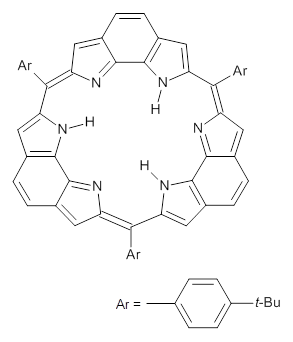
References
(1) Fukuzumi, S.; Ohkubo, K.; Ishida, M.; Preihs, C.; Chen, B.; Borden, W. T.; Kim, D.; Sessler, J. L. "Formation of Ground State Triplet Diradicals from Annulated Rosarin Derivatives by Triprotonation," J. Am. Chem. Soc. 2015, 137, 9780-9783, DOI: 10.1021/jacs.5b05309.
InChIs
1: InChI=1S/C45H24N6/c1-2-8-29-28(7-1)34-16-22-13-24-18-36-30-9-3-4-10-31(30)38-20-26(50-43(38)42(36)48-24)15-27-21-39-33-12-6-5-11-32(33)37-19-25(49-44(37)45(39)51-27)14-23-17-35(29)41(47-23)40(34)46-22/h1-21,46,49-50H/b22-13-,23-14-,24-13-,25-14-,26-15-,27-15-
InChIKey=UMEHBSBBAUKCTH-UIJINECUSA-N