Inverted carbon atoms, where the bonds from a single carbon atom are made to four other atoms which all on one side of a plane, remain a subject of fascination for organic chemists. We simply like to put carbon into unusual environments! Bremer, Fokin, and Schreiner have examined a selection of molecules possessing inverted carbon atoms and highlights some problems both with experiments and computations.1
The prototype of the inverted carbon is propellane 1. The Cinv-Cinv bond distance is 1.594 Å as determined in a gas-phase electron diffraction experiment.2 A selection of bond distance computed with various methods is shown in Figure 1. Note that CASPT2/6-31G(d), CCSD(t)/cc-pVTZ and MP2 does a very fine job in predicting the structure. However, a selection of DFT methods predict a distance that is too short, and these methods include functionals that include dispersion corrections or have been designed to account for medium-range electron correlation.
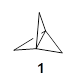
|
|
|
CASPT2/6-31G(d) CCSD(T)/cc-pVTZ MP2/cc-pVTZ MP2/cc-pVQZ B3LYP/6-311+G(d,p) B3LYP-D3BJ/6-311+G(d,p) M06-2x/6-311+G(d,p) |
1.596 1.595 1.596 1.590 1.575 1.575 1.550 |
Figure 1. Optimized Structure of 1 at MP2/cc-pVTZ, along with Cinv-Cinv distances (Å) computed with different methods.
Propellanes without an inverted carbon, like 2, are properly described by these DFT methods; the C-C distance predicted by the DFT methods is close to that predicted by the post-HF methods.
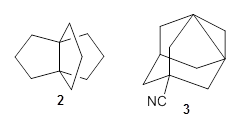
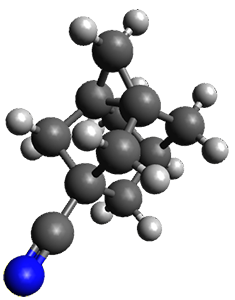
The propellane 3 has been referred to many times for its seemingly very long Cinv-Cinv bond: an x-ray study from 1973 indicates it is 1.643 Å.3 However, this distance is computed at MP2/cc-pVTZ to be considerably shorter: 1.571 Å (Figure 2). Bremer, Fokin, and Schreiner resynthesized 3 and conducted a new x-ray study, and find that the Cinv-Cinv distance is 1.5838 Å, in reasonable agreement with the computation. This is yet another example of where computation has pointed towards experimental errors in chemical structure.
|
Figure 2. MP2/cc-pVTZ optimized structure of 3.
However, DFT methods fail to properly predict the Cinv-Cinv distance in 3. The functionals B3LYP, B3LYP-D3BJ and M06-2x (with the cc-pVTZ basis set) predict a distance of 1.560, 1.555, and 1.545 Å, respectively. Bremer, Folkin and Schreiner did not consider the ωB97X-D functional, so I optimized the structure of 3 at ωB97X-D/cc-pVTZ and the distance is 1.546 Å.
Inverted carbon atoms appear to be a significant challenge for DFT methods.
References
(1) Bremer, M.; Untenecker, H.; Gunchenko, P. A.; Fokin, A. A.; Schreiner, P. R. "Inverted Carbon Geometries: Challenges to Experiment and Theory," J. Org. Chem. 2015, 80, 6520–6524, DOI: 10.1021/acs.joc.5b00845.
(2) Hedberg, L.; Hedberg, K. "The molecular structure of gaseous [1.1.1]propellane: an electron-diffraction investigation," J. Am. Chem. Soc. 1985, 107, 7257-7260, DOI: 10.1021/ja00311a004.
(3) Gibbons, C. S.; Trotter, J. "Crystal Structure of 1-Cyanotetracyclo[3.3.1.13,7.03,7]decane," Can. J. Chem. 1973, 51, 87-91, DOI: 10.1139/v73-012.
InChIs
1: InChI=1S/C5H6/c1-4-2-5(1,4)3-4/h1-3H2
InChIKey=ZTXSPLGEGCABFL-UHFFFAOYSA-N
3: InChI=1S/C11H13N/c12-7-9-1-8-2-10(4-9)6-11(10,3-8)5-9/h8H,1-6H2
InChIKey=KTXBGPGYWQAZAS-UHFFFAOYSA-N

Steven Bachrach responded on 07 Jul 2015 at 7:13 am #
I was informed today that I overlooked a senior author on this paper, Matthias Bremer. I have edited the post to include his name as well, and I apologize for this oversight.
Henry Rzepa responded on 08 Jul 2015 at 10:11 am #
Inverted carbons can be considered as exotic species, for which the regular single reference determinant methods may be a greater approximation than normal. I would be more convinced if multi-reference or VB methods were applied to such systems and compared with experiment (measured at low temperatures, say 90K).
Matthias Bremer responded on 10 Jul 2015 at 3:21 pm #
For [1.1.1]propellane the singlet-triplet gap is 79 kcal/mol (D. Feller, E. R. Davidson, J. Am. Chem. Soc. 1987, 109, 4133.). For 1,3-didehydro-5-cyanoadamantane we calculate 37.5 kcal/mol at M06-2X/cc-pVTZ. Higher spin states are obviously not (very) relevant in these propellane-type structures.
Determining the X-ray crystal structure at even lower temperatures would probably shorten the C-C distance even more and bring it closer to the MP2-calculated value.
Henry Rzepa responded on 13 Jul 2015 at 11:36 pm #
Yes, that singlet-triplet gap is pretty definitive.
I also looked up the temperature of the crystal structure determination, 200K. It is perfectly feasible to do these at 140K as standard or even 90K as non-standard nowadays.
I would also make the case for more determinations using high angle scattering methods (doi: 10.1002/anie.200500169) to measure the density in the region of bonds rather than just for the inner shells of atoms. This would allow e.g. QTAIM analysis to probe the properties of the density in the region of interesting or controversial bonds. Synchrotron sources are certainly available in e.g. Germany; perhaps its the quality/size of the crystal required that inhibits more such experimental bonding information from emerging in quantity?
Matthias Bremer responded on 14 Jul 2015 at 1:41 am #
Propellanes have been studied by high precision X-ray diffraction and Bader-Analysis. For example see: M. Messerschmidt, S. Scheins, L. Grubert, M. Patzel, G. Szeimies, C. Paulmann, P. Luger, Angew. Chem. 2005, 44, 3925-3928. (doi: 10.1002/anie.200500169).
Matthias Bremer responded on 14 Jul 2015 at 2:01 am #
Sorry, I just saw (from the DOI!) that Henry Rzepa and I mentioned the same paper by Luger et al. The bonding situation in propellanes has been extensively studied and we cite the pertinent literature. We show that DFT appears to have a problem with this very special kind of bonding. As experimentalists, we are unable to offer an explanation, but would certainly like to encourage a more profound theoretical study.
Ragnar responded on 15 Jul 2015 at 8:32 am #
Very interesting systems. Just tried a few ORCA calculations (def2-TZVP basis) on propellane 1. It seems that the DFT functionals struggling has a lot to do with the balance between HF exchange and correlation.
Method Bond distance
HF 1.5409
MP2 1.5939
BLYP 1.5905
B3LYP 1.5669
CAM-B3LYP 1.5460
BHLYP 1.5451
B2PLYP 1.5776
DSD-BLYP-D3 1.5788
DSD-PBEP86-D3 1.5812
LC-BLYP 1.5334
OLYP 1.5806
revTPSS 1.5829
The global (B3LYP,BHLYP)and range-separated hybrids (LC-BLYP, CAM-B3LYP) all inherit the bad behaviour of the HF method it seems. It takes MP2 correlation to make this right as seen in the MP2 method and to a lesser extent in the double-hybrids (B2PLYP and DSD methods) that include a fraction of MP2 correlation.
Interestingly, the non-hybrid GGA (BLYP, OLYP) and meta-GGA functionals (revTPSS) are much closer to experiment and CCSD(T) results.
Henry Rzepa responded on 16 Jul 2015 at 4:35 am #
Ragnar;
Those are useful calibrations. Thanks!
On another blog, three of us are discussing issues with the wavefunctions of molecules known as electrides, where calculations done by one of us are being analysed by the others. To exchange the data, we are using eg zenodo.org as a sort of dropbox, albeit one run by CERN (of LHC-Higgs particle fame) rather than a commercial organisation, and where additionally one receives a DOI for the dataset (with all that implies, in particular standard dissemination of metadata describing the dataset).
Would you consider eg putting the ORCA outputs of the calculations above into such an archive, and send us its DOI? This would be a manual deposition, but it is perfectly possible to automate the process as well. An automated system we developed and use for this purpose is also archived at Zenodo, as DOI: 10.5281/zenodo.19174.
Might I also follow this up with a general comment that the use of the ORCID researcher ID system is rapidly gathering pace, and I think can be usefully used on eg blogs to help provide information about participants.