A recent reinvestigation of the structure of 2-oxazoline demonstrates the difficulties that many computational methods can still have in predicting structure.
Samdal, et al. report the careful examination of the microwave spectrum of 2-oxzoline and find that the molecule is puckered in the ground state.1 It’s not puckered by much, and the barrier for inversion of the pucker, through a planar transition state is only 49 ± 8 J mol-1. The lowest vibrational frequency in the non-planar ground state, which corresponds to the puckering vibration, has a frequency of 92 ± 15 cm-1. This low barrier is a great test case for quantum mechanical methodologies.
And the outcome here is not particularly good. HF/cc-pVQZ, M06-2X/cc-pVQZ, and B3LYP/cc-pVQZ all predict that 2-oxazoline is planar. More concerning is that CCSD and CCSD(T) with either the cc-pVTZ or cc-pVQZ basis sets also predict a planar structure. CCSD(T)-F12 with the cc-pVDZ predicts a non-planar ground state with a barrier of only 8.5 J mol-1, but this barrier shrinks to 5.5 J mol-1 with the larger cc-pVTZ basis set.
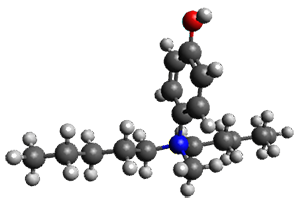
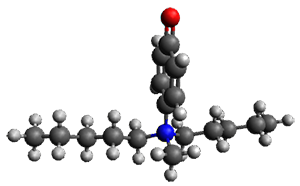
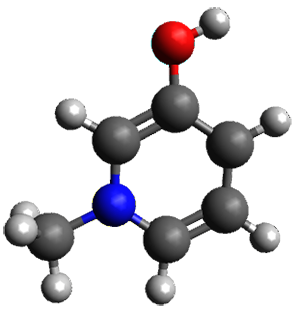
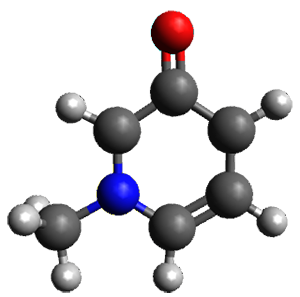
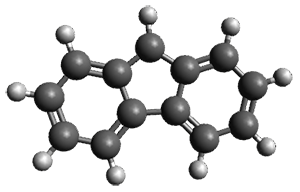
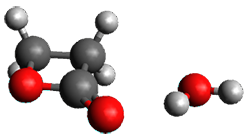
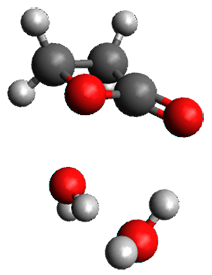
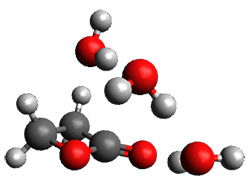
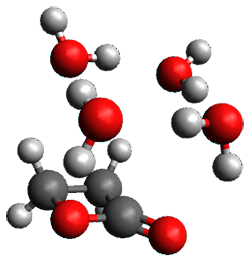
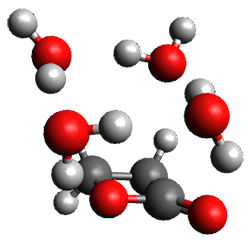
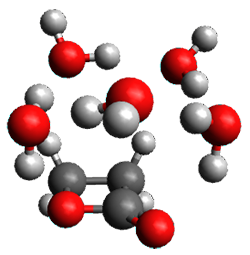
The only method that has good agreement with experiment is MP2. This method predicts a non-planar ground state with a pucker barrier of 11 J mol-1 with cc-pVTZ, 39.6 J mol-1 with cc-pVQZ, and 61 J mol-1 with the cc-pV5Z basis set. The non-planar ground state and the planar transition state of 2-oxazoline are shown in Figure 1. The computed puckering vibrational frequency does not reproduce the experiment as well; at MP2/cc-pV5Z the predicted frequency is 61 cm-1 which lies outside of the error range of the experimental value.
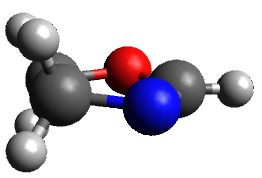
Non-planar |
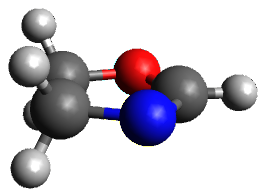
Planar TS |
Figure 1. MP2/cc-pV5Z optimized geometry of the non-planar ground state and the planar transition
state of 2-oxazoline.
References
(1) Samdal, S.; Møllendal, H.; Reine, S.; Guillemin, J.-C. "Ring Planarity Problem of 2-Oxazoline Revisited Using Microwave Spectroscopy and Quantum Chemical Calculations," J. Phys. Chem. A 2015, 119, 4875–4884, DOI: 10.1021/acs.jpca.5b02528.
InChIs
2-oxazoline: InChI=1S/C3H5NO/c1-2-5-3-4-1/h3H,1-2H2
InChIKey=IMSODMZESSGVBE-UHFFFAOYSA-N