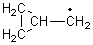The very simple carbene hydroxymethylene, HOCH, has finally been prepared and characterized.1 Glyoxylic acid CHOCO2H is subjected to high-vacuum laser photolysis. It fragments into HOCH, which is then trapped into an argon matrix. The experimental IR frequencies match up very well with the CCSD(T)/cc-pVQZ harmonic frequencies of the trans isomer 1t that are also adjusted for anharmonic effects. The computed vertical excitation energy of 415 nm matches well with the experimental value of the maximum absorption in the UV/vis spectra of 427 nm.
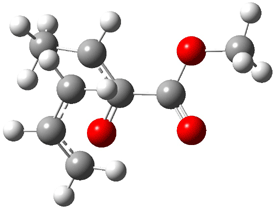
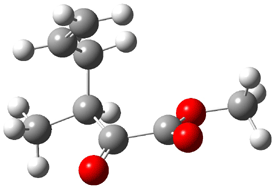
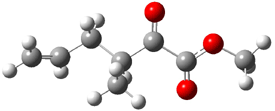
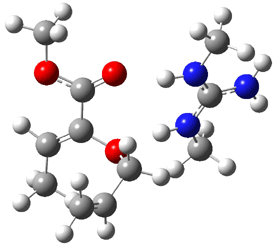
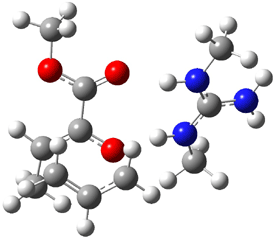
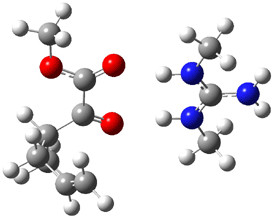
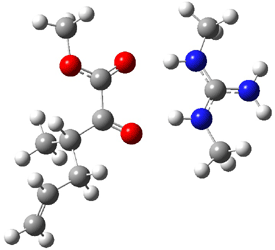
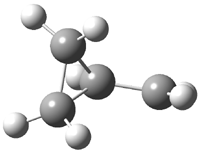
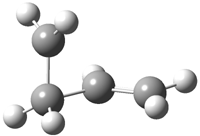
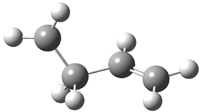
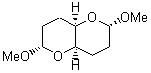
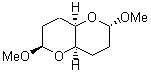
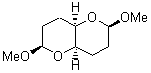
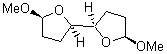
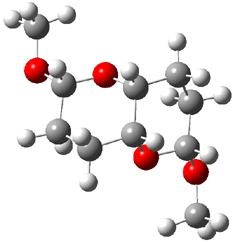
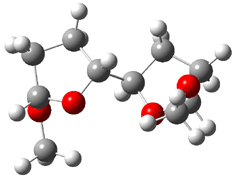
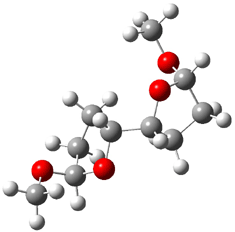
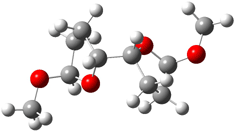
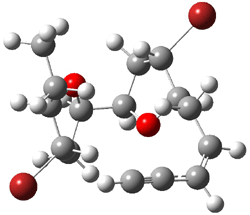
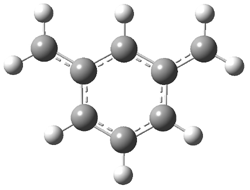
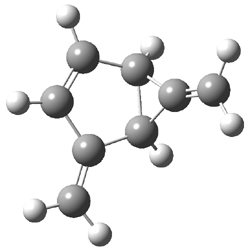
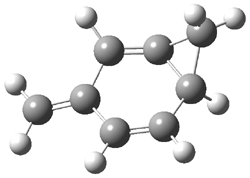
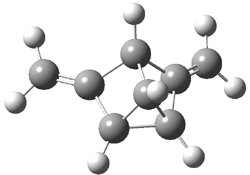
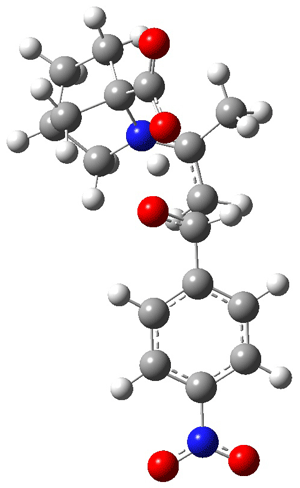
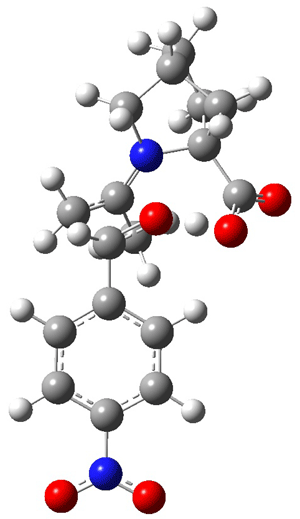
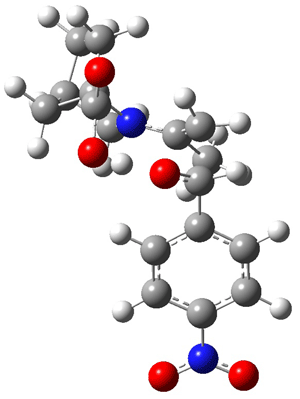
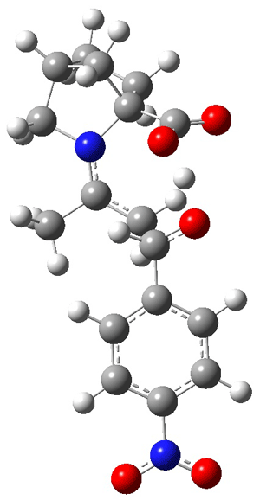
The other very interesting experimental result is that HOCH has a lifetime of about 2 hours in the matrix, while the deuterated species DOCH is stable. To explain these results, Schreiner, Allen and co-workers optimized a number of structures on the PES at CCSD(T)/cc-pVQZ and computed their energies using the focal point technique. The optimized structures and their relative energies are given in Figure 1.
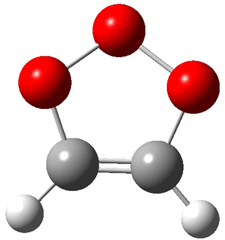
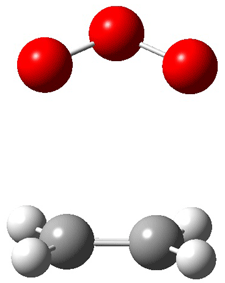
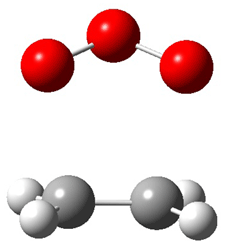
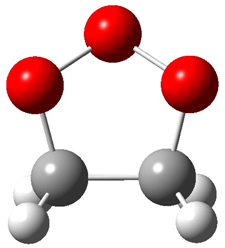
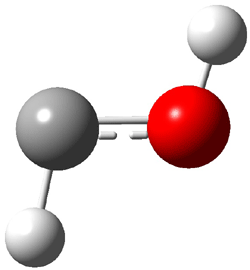 1t (0.0) |
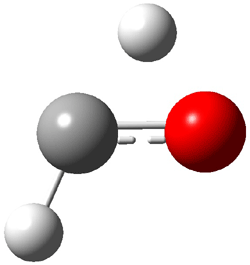 TS2 (29.7) |
 2 (-52.1) |
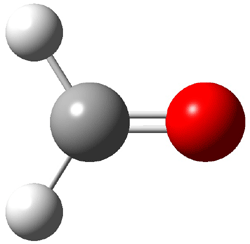
 TS1(26.8) |
|
|
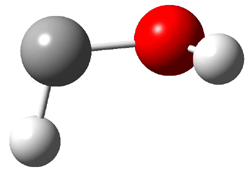
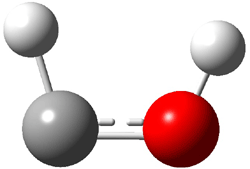 1c (4.4) |
|
|
Figure 1. Optimized CCSD(T)/cc-pVQZ structures of HOCH isomers and their Focal Point relative energies (kcal mol-1).1
The barriers for rearrangement from 1t are both very high. Rearrangement to formaldehyde 2 requires crossing a barrier of 29.7 kcal mol-1, while the barrier to convert to the cis isomer 1c is 26.8 kcal mol-1. (Note that from 1c a cleavage into CO and H2 can occur, but this barrier is another 47.0 kcal mol-1.) These barriers are too large to be crossed at the very low temperatures of the matrices. However, using the intrinsic reaction potential at CCSD(T)/cc-pVQZ and WKB theory, the tunneling lifetime of HOCH is computed to be 122 minutes, in excellent accord with the experiment. The lifetime for DOCH is computed to be over 1200 years. Thus, the degradation of hydroxymethylene is entirely due to tunneling through a very large classical barrier! This rapid tunneling casts serious doubt on the ability to ever identify any hydroxymethylene in interstellar space.
References
(1) Schreiner, P. R.; Reisenauer, H. P.; Pickard IV, F. C.; Simmonett, A. C.; Allen, W.
D.; Matyus, E.; Csaszar, A. G., "Capture of hydroxymethylene and its fast disappearance through tunnelling," Nature, 2008, 453, 906-909, DOI: 10.1038/nature07010.
InChI
1: InChI=1/CH2O/c1-2/h1-2H
2: InChI=1/CH2O/c1-2/h1H2