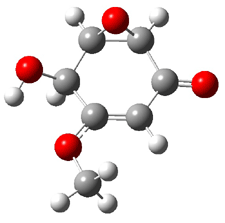
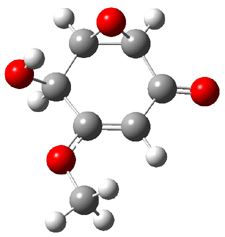
I discuss computational prediction of optical rotation in Chapter 1.6.3. I want to address a new protocol for determining absolute configuration using computed spectral properties and a recent review of state-of-the-art computational methods for predicting optical activity.
Stephens and the Gaussian personnel developed the techniques for computing optical rotation, electronic circular dichroism (ECD) and vibrational circular dichroism (VCD).1-4 Over the past year, Stephens has implemented a protocol for computing these properties in order to determine the absolute configuration of a chiral molecule.5-7 The first application was to the structures of the related sesquiterpenes 1-4.5 A Monte Carlo conformational search is first carried out using MMFF94, and all low energy conformers are reoptimized at B3LYP/6-31G*. Restricted searches by varying some dihedral angles are also sometimes used to insure that all reasonable low-energy conformations have been identified. Then, using these optimized geometries,specific rotations and ECD are computed at TDDFT/B3LYP/aug-cc-pVDZ, and IR and VCCD spectra computed at B3LYP and B3PW91 with the TZ2P basis set.

The computed and experimental optical rotations at the sodium D line for 1-4 are listed in Table 1. The computed values of [α]D for 1 as the 1R,2R,5S,8R,11R isomer (as shown above) is in reasonable agreement with the experimental value – with error similar to those I describe in the book. This is in agreement with the assigned absolute configuration of naturally occurring 1. While the core structures of 2-4 are likely to be identical to 1 assuming similar biosynthesis, their absolute configurations have not been determined. The computed optical rotation for 3 and 4 are again in reasonable agreement with experiment, but for 2 the errors are large for either enantiomer. This is where ECD and VCD are valuable. The computed ECD and VCD spectra of 1 are in extraordinary agreement with the experimental spectra, confirming the assigned absolute configuration. Stephens reports the ECD and VCD spectra of 2-4 and finds that they all have identical configurationas of the core. He suggests that experimental determination of these VCD spectra will confirm all of their absolute configurations.
Table 1. Experimental and computed optical rotation for 1-4 at the sodium D line.
|
|
|
[α]D (expt)
|
[α]D (calc)
|
|
1
|
-52.7,a -60.5,b -44.6c
|
-38.8c
|
|
2
|
55.7d
|
10.5c
|
|
3
|
-60e
|
-20.6c
|
|
4
|
-110e
|
-220.8c
|
|
aRef. 8. bRef. 9. cRef. 10. eRef. 5. eRef. 11
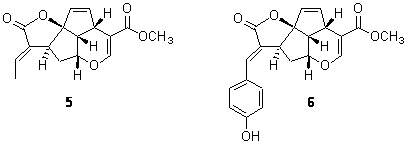
A second study involved the iridoids 5 and 6.7 Plumericin 5 has the absolute configuration shown below with [α]D = +204. The more recently discovered prismatomerin 6 has [α]D = -136, suggesting that the core polycyclic portion may have opposite absolute configuration. Stephens prepared the acetate of 6 and experimentally determined its VCD spectrum. The VCD spectrum was then computed using the above protocol. The computed spectrum for the enantiomer with the same absolute configuration as 5 matches the experimental spectrum. Thus, 5 and 6 have the same absolute configuration. Stephens concludes with the warning that optical activity of analogous compounds can be quite different and is not suitable for obtaining configuration information. Rather, VCD is a much more suitable test, especially when experimental and computed spectra are utilized.
I will finish this post with a brief recap of the optical rotation computations a few of the molecules discussed in a recent review by Crawford.12 Crawford implements a linear-response coupled clusters with modified velocity gauge protocol. He compares the optical rotation computed with this method, the time-dependent DFT approach developed by Stephens et al and experiment. He describes systems where the CC approach performs much better than DFT, where DFT performs better than CC, but for the wrong reason, and a case where DFT appears to perform better than CC.
The optical rotation of (P)-(+)-[4]-triangulane 7 at a variety of wavelengths has been determined both experimentally and computationally. These results are listed in Table 2. It is readily apparent that CCSD performs much better than DFT. The poor performance of the DFT method is linked to electronic excitation energies that are too small.

Table 2. Optical rotation of 7.
|
|
Wavelength (nm)
|
B3LYPa
|
CCSD(MVG)a
|
Expt.b
|
|
589
|
221.5
|
196.0
|
192.7
|
|
578
|
231.4
|
204.5
|
201.3
|
|
546
|
264.3
|
232.9
|
229.7
|
|
436
|
460.7
|
398.7
|
400.2
|
|
365
|
752.2
|
635.4
|
648.2
|
|
aRef. 13. bRef. 14.
The ORD of (S)-methyloxirane show a change of sign: -8.39 at 633 nm and +7.39 at 355 nm. CCSD predicts a reasonable value at 633 nm but gets the wrong sign at the shorter wavelength. On the other hand, B3LYP does predict the sign change. However, this seemingly correct result is due to (once again) underestimation of the excitation energy.
Lastly, Crawford reports on his study of (1S,4S)-norbornenone. The optical rotation of the sodium D line is -1146, and B3LYP does a very reasonable job in predicting a value of -1214. However, CCSD(MVG) grossly underestimates this value at -558. Though B3LYP again underestimates the excitation energy it appears to get the energy and rotational strength near the liquid-phase values. Most worrisome is that Crawford discounts basis set improvements and higher order correlation effects, and holds some hope for a significant difference in gas-phase vs solution phase rotations.
References
(1) Stephens, P. J., "Theory of Vibrational Circular Dichroism," J. Phys. Chem. 1985, 89, 748-752, DOI: 10.1021/j100251a006.
(2) Cheeseman, J. R.; Frisch, M. J.; Devlin, F. J.; Stephens, P. J., "Hartree-Fock
and Density Functional Theory ab Initio Calculation of Optical Rotation Using GIAOs: Basis Set
Dependence," J. Phys. Chem. A, 2000, 104, 1039-1046, DOI: 10.1021/jp993424s.
(3) Stephens, P. J.; Devlin, F. J.; Cheeseman,J. R.; Frisch, M. J., "Calculation of Optical Rotation Using Density Functional Theory," J. Phys. Chem. A, 2001, 105, 5356-5371, DOI: 10.1021/jp0105138.
(4) Stephens, P. J.; McCann, D. M.; Cheeseman, J. R.; Frisch, M. J., "Determination of absolute configurations of chiral molecules using ab initio time-dependent Density Functional Theory calculations of optical rotation: How reliable are absolute configurations obtained for molecules with small rotations?," Chirality, 2005, 17, S52-S64, DOI: 10.1002/chir.20109.
(5) Stephens, P. J.; McCann, D. M.; Devlin, F. J.; Smith, A. B., "Determination of the Absolute Configurations of Natural Products via Density Functional Theory Calculations of Optical Rotation, Electronic Circular Dichroism, and Vibrational Circular Dichroism: The Cytotoxic Sesquiterpene Natural Products Quadrone, Suberosenone, Suberosanone, and Suberosenol A Acetate," J. Nat. Prod., 2006, 69, 1055-1064, DOI: 10.1021/np060112p.
(6) Stephens, P. J.; Pan, J. J.; Devlin, F. J.; Urbanova, M.; Hajicek, J., "Determination of the Absolute Configurations of Natural Products via Density Functional Theory Calculations of Vibrational Circular Dichroism, Electronic Circular Dichroism and Optical Rotation: The Schizozygane Alkaloid Schizozygine," J. Org. Chem., 2007, 72, 2508-2524, DOI: http://dx.doi.org/10.1021/jo062567p.
(7) Stephens, P. J.; Pan, J. J.; Krohn, K., "Determination of the Absolute Configurations of Pharmacological Natural Products via Density Functional Theory Calculations of Vibrational
Circular Dichroism: The New Cytotoxic Iridoid Prismatomerin," J. Org. Chem., 2007, 72, 7641-7649, DOI: 10.1021/jo071183b.
(8) Smith, A. B.; Konopelski, J. P.; Wexler, B. A.; Sprengeler, P. A., "Quadrone structural and synthetic studies. Total synthesis of natural (-)-quadrone, the (+)-enantiomer, and the racemate. Conformational analysis, circular dichroism, and determination of absolute stereochemistry," J. Am. Chem. Soc., 1991, 113, 3533-3542, DOI: 10.1021/ja00009a047.
(9) Wijeratne, E. M. K.; Turbyville, T. J.; Zhang, Z.; Bigelow, D.; Pierson, L. S.; VanEtten, H. D.; Whitesell, L.; Canfield, L. M.; Gunatilaka, A. A. L., "Cytotoxic Constituents of Aspergillus terreus from the Rhizosphere of Opuntia versicolor of the Sonoran Desert," J. Nat. Prod., 2003, 66, 1567-1573, DOI: 10.1021/np030266u.
(10) Bokesch, H. R.; McKee, T. C.; Cardellina II, J. H.; Boyd, M. R., "Suberosenone, a new cytotoxin from Subergorgia suberosa," Tetrahedron Lett., 1996, 37, 3259-3262, DOI: 10.1016/0040-4039(96)00528-X
(11) Sheu, J. H.; Hung, K. C.; Wang, G. H.; Duh, C. Y., "New Cytotoxic Sesquiterpenes from the Gorgonian Isis hippuris," J. Nat. Prod., 2000, 63, 1603-1607, DOI: 10.1021/np000271n.
(12) Crawford, T. D.; Tam, M. C.; Abrams, M. L., "The Current State of Ab Initio Calculations of Optical Rotation and Electronic Circular Dichroism Spectra," J. Phys. Chem. A, 2007, 111, 12057-12068, DOI: 10.1021/jp075046u.
(13) Crawford, T. D.; Owens, L. S.; Tam, M. C.; Schreiner, P. R.; Koch, H., "Ab Initio Calculation of Optical Rotation in (P&)-(+)-[4]Triangulane," J. Am. Chem. Soc., 2005, 127, 1368-1369, DOI: 10.1021/ja042787p.
(14) de Meijere, A.; Khlebnikov, A. F.; Kozhushkov, S. I.; Kostikov, R. R.; Schreiner, P. R.; Wittkopp, A.; Rinderspacher, C.; Menzel, H.; Yufit, D. S.; Howard, J. A. K., "The First Enantiomerically Pure [n]Triangulanes and Analogues: σ-[n]Helicenes with Remarkable Features," Chem. Eur. J., 2002, 8, 828-842, DOI: 10.1002/1521-3765(20020215)8:4<828::AID-CHEM828>3.0.CO;2-Y
InChIs
1: InChI=1/C15H20O3/c1-14(2)7-15-9-4-3-8(14)10(15)5-12(16)11(15)6-18-13(9)17/h8-11H,3-7H2,1-2H3/t8?,9-,10?,11?,15?/m1/s1 PubChem
InChIKey: BBIDMUQZCCGABN-UDZYVRSQBU
2: InChI=1/C15H22O/c1-9-5-6-11-12-7-13(16)10(2)15(9,12)8-14(11,3)4/h9,11-12H,2,5-8H2,1,3-4H3/t9-,11?,12?,15?/m0/s1
InChIKey: JDGYVUJBJYXKSX-NCLPGTSEBA
3: InChI=1/C15H24O/c1-9-5-6-11-12-7-13(16)10(2)15(9,12)8-14(11,3)4/h9-12H,5-8H2,1-4H3/t9-,10+,11?,12?,15?/m0/s1
InChIKey: KHINYKJYBNWSSP-BIGXPMCQBN
4: InChI=1/C17H26O2/c1-10-6-7-13-14-8-15(19-12(3)18)11(2)17(10,14)9-16(13,4)5/h10,13-15H,2,6-9H2,1,3-5H3/t10-,13?,14?,15+,17?/m0/s1
InChIKey: UZRAQUNNGNYEHD-XDUGHSHMBF
5: InChI=1/C16H18O5/c1-3-8-11-6-12-13-9(10(7-20-12)14(17)19-2)4-5-16(11,13)21-15(8)18/h3,7,9,11-13H,4-6H2,1-2H3/b8-3+/t9-,11-,12-,13-,16-/m1/s1
InChIKey: QOWSZGWHIKPQIA-WCDIAXTGBA
PubChem
6: InChI=1/C21H20O6/c1-25-19(23)15-10-26-17-9-16-14(8-11-2-4-12(22)5-3-11)20(24)27-21(16)7-6-13(15)18(17)21/h2-5,8,10,13,16-18,22H,6-7,9H2,1H3/b14-8+/t13-,16-,17-,18-,21-/m1/s1
InChIKey: OXYVEVVOLQYXPZ-SOSYHPOKBY
7: InChI=1/C9H12/c1-2-7(1)5-9(7)6-8(9)3-4-8/h1-6H2
InChIKey: JCHCWAJGYWGXMQ-UHFFFAOYAV