It is striking to me that the absolute configuration of relatively simple compounds remains problematic even today. The structure of two naturally-occurring phytotoxic enantiomers 1, called regiolone and isosclerone, are finally definitively defined using a computational approach.
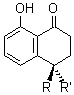
(R)-1: R = OH, R’ = H
(S)-1: R = H, R’ = OH
Isosclerone is the dextrorotatory isomer, while regiolone is the levorotatory isomer. The question though is which one is R and which one is S? Evidente and co-workers arbitrarily decided to compute the spectral properties of the S isomer.1 They located four low energy conformers at B3LYP/6-31G* and B3LYP/TZVP. (These conformers are not shown here as the authors did not deposit the coordinates. Reviewers and editors – please insist that this computational data be mandatory for publication!) The conformer relative energies, listed in Table 1, are dependent on the method, however, the two lowest energy structures will dominate the population and both will be present to a significant extent, regardless of which energy set is used. The optical rotation [α]D was computed at B3LYP/6-31G*//B3LYP/TZVP, and these too are listed in Table 1. The Boltzmann-weighted [α]D is 21.8. Even though the lowest energy conformer contributes a negative rotation, the much larger positive rotation due to the second-lowest energy conformer, along with the two other conformers, will dominate to dictate the OR value. This suggests that the enantiomers are (S)(+)-1 and (R)(-)-1. Computed ECD spectra confirm this assignment; the computed ECD of the (S) isomer is a near mirror image of the experimental ECD of the (-)-1 compound. Therefore, regiolone is (R)(-)-1 and isosclerone is (S)(+)-1.
Table 1. Relative free energies (kcal mol-1) and [α]D of the conformers of (S)-1.a
|
conformer |
ΔG, 6-31G* |
ΔG, TZVP |
[α]Db |
| A | 0.43 | 0.0 | -17.50 |
| B | 0.0 | 0.32 | 67.92 |
| C | 1.21 | 1.03 | 95.72 |
| D | 1.84 | 1.48 | 17.72 |
aAll computations performed with B3LYP. bAt B3LYP/6-31G*//B3LYP/TZVP
References
(1) Evidente, A.; Superchi, S.; Cimmino, A.; Mazzeo, G.; Mugnai, L.; Rubiales, D.; Andolfi, A.; Villegas-Fernández, A. M., "Regiolone and Isosclerone, Two Enantiomeric Phytotoxic Naphthalenone Pentaketides: Computational Assignment of Absolute Configuration and Its Relationship with Phytotoxic Activity," Eur. J. Org. Chem., 2011, 5564-5570, DOI: 10.1002/ejoc.201100941
InChIs
Regiolone: InChI=1/C10H10O3/c11-7-4-5-9(13)10-6(7)2-1-3-8(10)12/h1-3,7,11-12H,4-5H2/t7-/m1/s1
InChIKey=ZXYYTDCENDYKBR-SSDOTTSWBB
Isosclerone: InChI=1/C10H10O3/c11-7-4-5-9(13)10-6(7)2-1-3-8(10)12/h1-3,7,11-12H,4-5H2/t7-/m0/s1
InChIKey=ZXYYTDCENDYKBR-ZETCQYMHBD

Dragon Eye Morrison responded on 15 Mar 2012 at 1:32 am #
Thanks for highlighting this paper – one that I probably wouldn’t have read otherwise.
This is a really nice, simple example of Boltzmann-weighted calculations – clearly very important here given the negative optical rotation of the global minimum.
I wanted to know your opinion on the missing coordinates in the SI. I often find similar omissions in computational papers and sometimes statements are made which cannot be supported by the data presented – it makes you start to question the validity of the results/statements (i.e. did the author simply neglect to publish the required information or did they make a false assumption).
Surely every computational chemist can easily recognise what information is useful to others in the community and necessary to support the thesis of a given paper. Is is simply laziness or do you think data is left out for reasons of self interest? (I don’t mean to criticise these authors, as I think it’s a more endemic problem – as you said it’s down to the reviewers/editors really).
Steven Bachrach responded on 15 Mar 2012 at 9:11 am #
I don’t think in most cases that there is anything nefarious or untoward going on. It is rather, I think, just a case of laziness or not knowing that supporting materials can be readily deposited in a somewhat more useful format than in the olden days, when it would go onto microfiche!
It is really the job of editors to rewrite the requirements for submission to make inclusion of this type of data mandatory for acceptance, just like the CIF file is required for all papers that report new crystal structures. In the computational world, at minimum the full optimized coordinates and the energies and the number of imaginary frequencies should be required. For experimental work, spectra should be required to characterize all new compounds. Ideally, these materials would not be deposited solely as pdf copies, but as files that can be readily reused!
Back in the early days of my blog (2007!) I would probably have re-optimized the structures reported in this paper and included them in the blog post – but I just don’t have the time to do this and I am simply more upset now that coordinates are not routinely deposited.