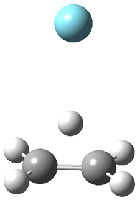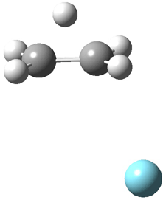Following on the great study of hydroxycarbene1 (see my blog post), Schreiner now reports on the synthesis and characterization of dihydroxycarbene 1.2 It is prepared by high-vacuum flash pyrolysis of oxalic acid (Scheme 1).
Scheme 1
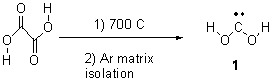
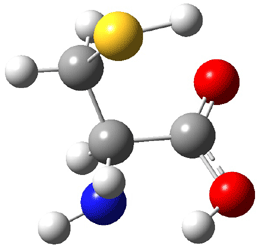
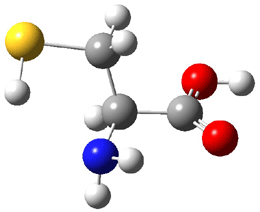
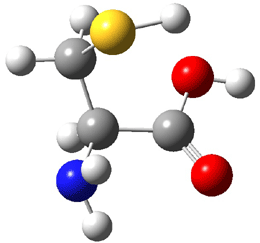
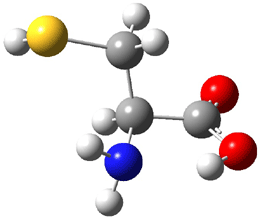
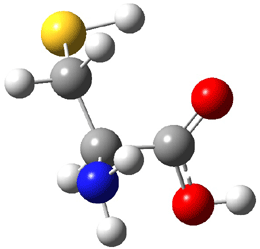
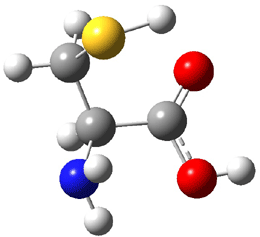
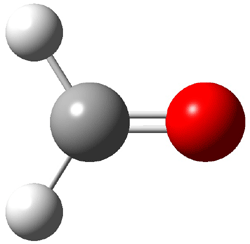
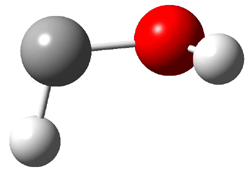
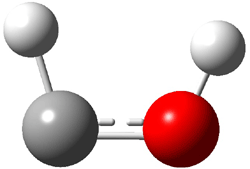
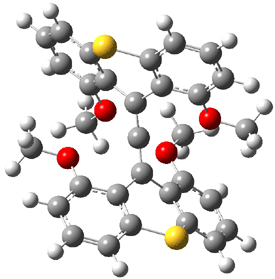
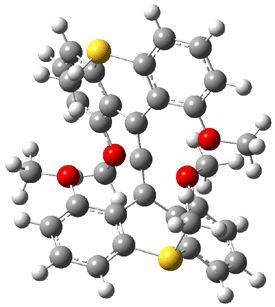
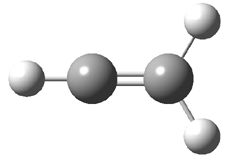
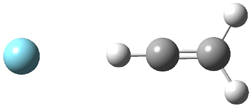
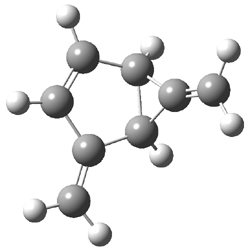
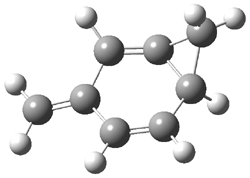
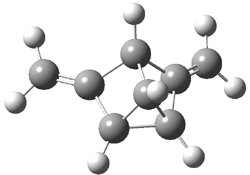
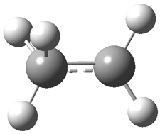
Dihydroxycarbene can exist in three different conformations characterized by the relationship about the C-O bond, either s-cis or s-trans. The three conformations are shown in Figure 1, and the s-trans,s-trans structure is the local energy minimum (computed at CCSD(T)/cc-pVTZ).
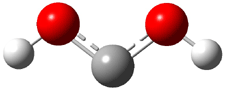 1tt (0.0) |
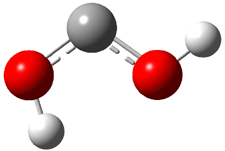 1ct (0.1) |
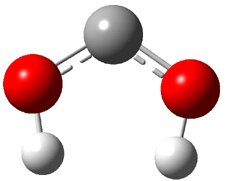 1cc (6.7) |
Figure 1. CCSD(T)/cc-pVTZ optimized geometries and relative energies (kcal mol-1) of the conformers of 1.2
Identification of the 1 is made through comparison of the experimental and computed IR vibrational frequencies. As an example, the experimental and computed frequencies for the s-trans,s-trans conformer are listed in Table 1. The agreement is excellent.
Table 1. Computed and experimental vibrational frequencies (cm-1) and intensities (in parentheses) of the s-trans,s-trans conformation of 1.2
|
|
||
|
vibration |
computed |
experiment |
|
1 |
3876.4 (23.5) |
3633.2 / 3628.6 (w) |
|
2 |
3871.4 (234.1) |
3625.1 (s) |
|
3 |
1443.1 (124.4) |
1386.2 (m) |
|
4 |
1370.5 (58.3) |
1289.0 / 1287.4 (w) |
|
5 |
1157.8 (470.6) |
1110.3 / 1109.3 (vs) |
|
6 |
1156.6 (1.4) |
|
|
7 |
742.4 (178.8) |
706.6 (s) |
|
8 |
672.4 (0.0) |
|
|
9 |
641.6 (11.2) |
|
|
|
||
Unlike hydroxycarbene, dihydroxycarbene is stable. The amazing instability of hydroxycarbene is due to tunneling through a large barrier: nearly 30 kcal mol-1.1 The tunneling route for the decomposition of 1 is more difficult for two reasons. First, its C-O bond is quite strong; the C-O distance is quite short, 1.325 Å. This makes a long distance that must be traversed in the tunneling mode. (The strong bond is due to π-donation from the oxygen lone pair into the empty carbon p orbital; this is noted by the large rotational barrier about the C-O bonds of 17 kcal mol-1!) Second, the activation barrier for decomposition is very high, at least 34 kcal mol-1.
References
(1) Schreiner, P. R.; Reisenauer, H. P.; Pickard Iv, F. C.; Simmonett, A. C.; Allen, W. D.; Matyus, E.; Csaszar, A. G., "Capture of hydroxymethylene and its fast disappearance through tunnelling," Nature, 2008, 453, 906-909, DOI: 10.1038/nature07010.
(2) Schreiner, P. R.; Reisenauer, H. P., "Spectroscopic Identification of Dihydroxycarbene13," Angew. Chem. Int. Ed., 2008, 47, 7071-7074, DOI: 10.1002/anie.200802105
InChIs
1: InChI=1/CH2O2/c2-1-3/h2-3H