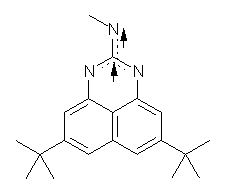
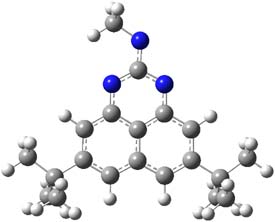
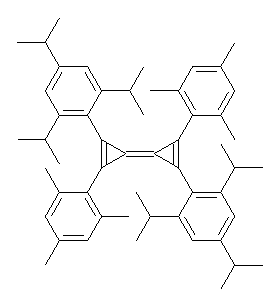
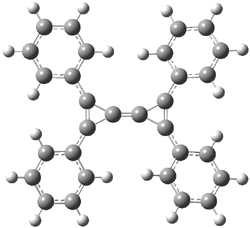
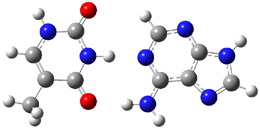
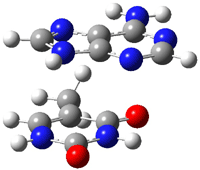
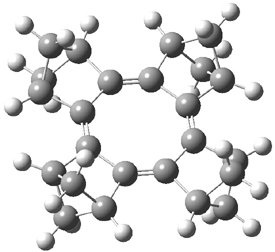
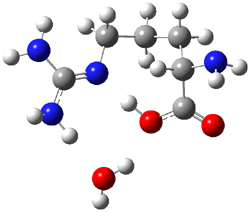
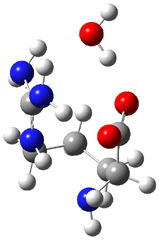
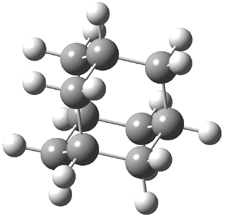
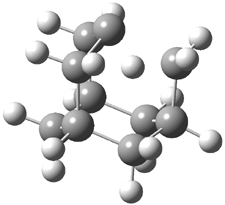
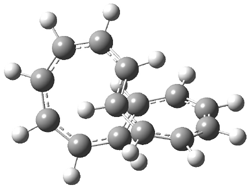
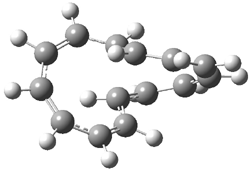
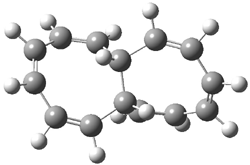
Here is another benchmark of the performance of DFT in handling difficult situations, in this case the interaction between nucleic acid base pairs. Sherrill1 has examined the 124 nucleic acid base pairs from the JSCH-2005 database2 compiled by Hobza and coworkers. This database includes 36 hydrogen bonded complexes, and example of which is shown in Figure 1a, and 54 stacked complex, one example of which is shown in Figure 1b.
Figure 1. Optimized geometries (RI-MP2/cc-pVTZ) of two representative structures of base pairs: (a) hydrogen bonded pair and (c) stacked pair.
The energies of these base pairs computed with four different functionals: PBE, PBE-D (where Grimme’s empirical dispersion correction3), and the recently developed MO5-2X4 and MO6-2X5 methods which attempt to treat mid-range electron correlation. The aug-cc-pVDZ basis set was used. These DFT energies are compared with the CCSD(T) energies of Hobza. The mean unsigned error (MUE) for the 28 hydrogen bonded complexes and the 54 stacked complexes are listed in Table 1.
Table 1. Mean unsigned error (kcal mol-1) of the four DFT
methods (relative to CCSD(T)) for the hydrogen bonded and stacked base pairs.
|
|
method
|
MUE (HB)
|
MUE (stacked)
|
|
|
PBE
|
2.59
|
7.57
|
|
PBE-D
|
0.70
|
1.53
|
|
MO5-2X
|
1.98
|
2.59
|
|
MO6-2X
|
1.62
|
1.08
|
|
A few interesting trends are readily apparent. First, PBE (representing standard GGA DFT methods) poorly describes the energy of the hydrogen bonded complexes, but utterly fails to treat the stacking interaction. Inclusion of the dispersion correction (PBE-D) results in excellent energies for the HB cases and quite reasonable results for the stacked pairs. Both of Truhlar’s functionals dramatically outperform PBE, though MO5-2X is probably still not appropriate for the stacked case. MO6-2X however seems to be a very reasonable functional for dealing with base pair interactions, indicating that mid-range correlation correction is sufficient to describe these complexes, and that the long-range correlation correction included in the dispersion correction, while giving improved results, is not essential.
References
(1) Hohenstein, E. G.; Chill, S. T.; Sherrill, C. D., "Assessment of the Performance of the M05-2X and M06-2X Exchange-Correlation Functionals for Noncovalent Interactions in Biomolecules," J. Chem. Theory Comput., 2008, 4, 1996-2000, DOI: 10.1021/ct800308k
(2) Jurecka, P.; Sponer, J.; Cerny, J.; P., H., "Benchmark database of accurate (MP2 and CCSD(T) complete basis set limit) interaction energies of small model complexes, DNA base pairs, and amino acid pairs," Phys. Chem. Chem. Phys., 2006, 8, 1985-1993, DOI: 10.1039/b600027d.
(3) Grimme, S., "Semiempirical GGA-type density functional constructed with a long-range dispersion correction," J. Comput. Chem., 2006, 27, 1787-1799, DOI: 10.1002/jcc.20495
(4) Zhao, Y.; Schultz, N. E.; Truhlar, D. G., "Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions," J. Chem. Theory Comput., 2006, 2, 364-382, DOI: 10.1021/ct0502763.
(5) Zhao, Y.; Truhlar, D. G., "The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals," Theor. Chem. Acc., 2008, 120, 215-241, DOI: 10.1007/s00214-007-0310-x.