Dihdroxycarbene was the subject of a post a few years ago relating to how this carbene does not undergo tunneling,1 while related hydroxycarbene do undergo a tunneling rearrangement.
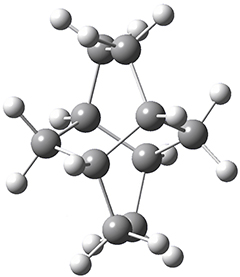
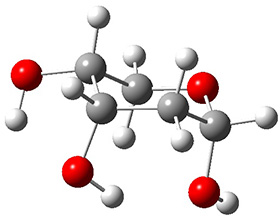
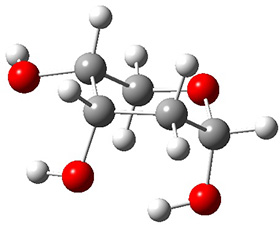
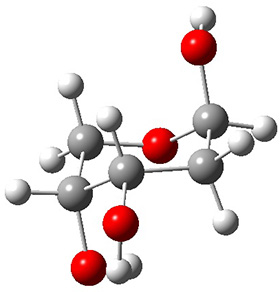
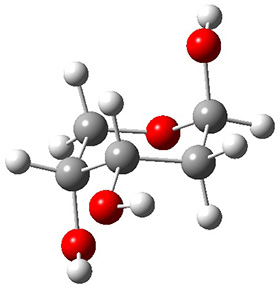
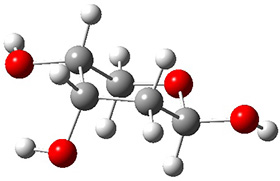
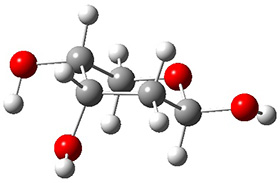
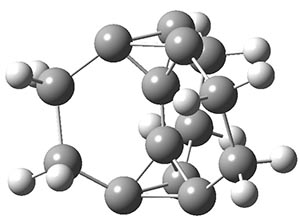
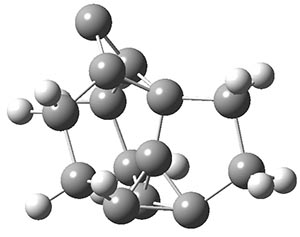
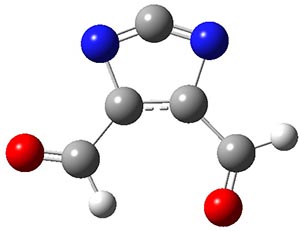
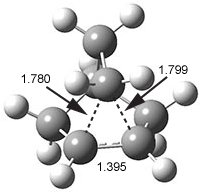
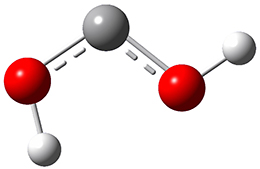
Now we have a gas-phase microwave determination of the trans,cis isomer of dihydroxycarbene.2 The computed CCSD(T)/cc-pCVQZ structure is shown in Figure 1. What is truly remarkable here is the amazing agreement between the experimental and computed structure – as seen in Table 1.The bond distance are in agreement within 0.001 Å and the bond angles agree within 0.3°! Just further evidence of the quality one can expect from high-level computations. And computing this structure was certainly far easier than the experiments!
|
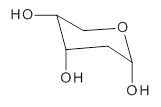
Figure 1. CCSD(T)/cc-pCVQZ optimized geometry of dihydroxycarbene.
Table 1. Experimental and computed (CCSD(T)/cc-pCVQZ) geometric parameters of dihydroxycarbene.a
|
|
||
|
|
Expt. |
Comp. |
|
C-O |
1.335 |
1.336 |
|
C-O |
1.309 |
1.309 |
|
O-Htrans |
0.961 |
0.960 |
|
O-Hcis |
0.976 |
0.975 |
|
O-C-O |
107.30 |
107.25 |
|
C-O-Htrans |
106.8 |
106.8 |
|
C-O-Hcis |
110.7 |
110.4 |
|
|
||
|
aDistances in Å and angles in deg. |
||
References
(1) Schreiner, P. R.; Reisenauer, H. P. "Spectroscopic Identification of Dihydroxycarbene," Angew. Chem. Int. Ed. 2008, 47, 7071-7074, DOI: 10.1002/anie.200802105.
(2) Womack, C. C.; Crabtree, K. N.; McCaslin, L.; Martinez, O.; Field, R. W.; Stanton, J. F.; McCarthy, M. C. "Gas-Phase Structure Determination of Dihydroxycarbene, One of the Smallest Stable Singlet Carbenes," Angew. Chem. Int. Ed. 2014, 53, 4089-4092, DOI: 10.1002/anie.201311082.
InChIs
Dihydroxycarbene: InChI=1S/CH2O2/c2-1-3/h2-3H
InChIKey=VZOMUUKAVRPMBY-UHFFFAOYSA-N