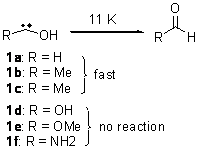
As we have noted in many previous posts, Schreiner has observed tunneling in hydroxycarbenes that is either very rapid (1a-c) or not at all (1d-f).1-4 In a recent paper his group investigates whether cyclopropylhydroxycarbene 2 might have an intermediate lifetime due to the π-donating effect of the three-member ring.5
Schreiner makes this carbene in his usual manner: flash pyrolysis of the cyclopropylglyoxylic acid. Let’s now consider three possible rearrangements of carbene 2. The hydrogen can migrate (Scheme 1, path a) to give cyclopropylcarboxyaldehyde 3 similar to what was observed with the related hydroxycarbenes. Carbon can migrate (Scheme 1, path b), opening up the three-member ring to give the cyclobutenol 4. This ring could open to the diene 5 and tautomerize to the ketone 6. Lastly, a hydrogen migration from carbon (Scheme 1, path c) would lead to 7. The relative energies of these species computed at CCSD(T)//cc-pVTZ//M06-2x//6-311++G(d,p) are shown in Scheme 1.
Scheme 1. Relative energies in kcal mol-1.
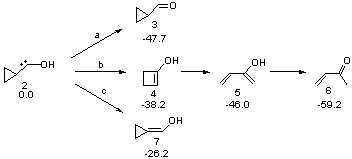
The computed barriers for the initial step of each pathway is +30.4 kcal mol-1 for path a, +21.9 kcal mol-1 for path b and +35.8 kcal mol-1 for path c. Thus, one might expect to see only the reaction along path b at low temperature and mostly along b at high temperature with some small percent along path a. So what actually occurs?
After capturing the flash pyrolysis product in an Ar matrix, besides the unreacted cyclopropylglyoxylic acid, 6, 3, and 2 are observed in an approximate 8:5:1 ratio. 2 is identified on the basis of the nice agreement between the experimental and computed IR frequencies. Irradiation of 2 in the matrix leads to clean conversion to 4, also identified by comparison of the observed and computed IR frequencies. This is all consistent with the computed activation barriers. In the pyrolysis, at high T, 6 is the major product and 3 is the minor product. At very low T (11 K), irradiation of 2 produces 4 (crossing only the lowest barrier) and not continuing further along the rearrangement path to 6.
What is perhaps most exciting is that 2 disappears slowly in the dark at both 11 K and 20 K, converting at the same rate to 3. The half life is 17.7 h, much longer than for the alkyl and aryl substituted hydroxycarbenes 1a-c. This confirms the stabilization effect of the cyclopropyl group, as does its large singlet-triplet gap. The computed tunneling half-life using the WKB approach is 16.6 h, in excellent agreement with experiment. And as expected for a tunneling phenomenon, the dueterated analog has a much longer half-life, computed to be 105 years. Experimentally, 2-d persists with no conversion to 3-d observed.
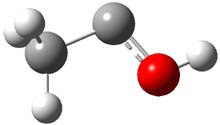
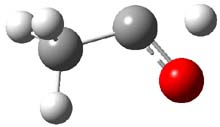
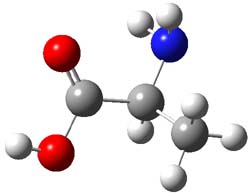
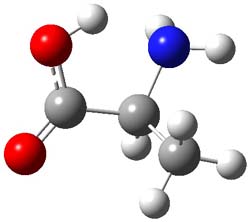
As with methylhydroxycarbene, we see here an example of tunneling control vs kinetic control. At high T, the reaction crosses the lowest barrier (shown in Figure 1a), proceeding to 4 and subsequent rearrangement products. At low T, the reaction crosses a higher barrier (shown in Figure 1b), but this path involves tunneling of the very light hydrogen atom only, producing 3.
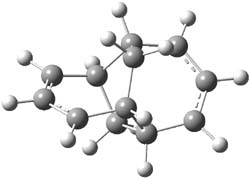
 TS 2 → 3 |
 TS 2 → 4 |
Figure 1. M06-2X/6-311++G(d,p) optimized geometry of the transition states connecting 2 to (a) 3 and (b) 4.
References
(1) Schreiner, P. R.; Reisenauer, H. P.; Pickard Iv, F. C.; Simmonett, A. C.; Allen, W. D.; Matyus, E.; Csaszar, A. G., "Capture of hydroxymethylene and its fast disappearance through tunnelling," Nature, 2008, 453, 906-909, DOI: 10.1038/nature07010.
(2) Schreiner, P. R.; Reisenauer, H. P., "Spectroscopic Identification of Dihydroxycarbene," Angew. Chem. Int. Ed., 2008, 47, 7071-7074, DOI: 10.1002/anie.200802105
(3) Gerbig, D.; Reisenauer, H. P.; Wu, C.-H.; Ley, D.; Allen, W. D.; Schreiner, P. R., "Phenylhydroxycarbene," J. Am. Chem. Soc., 2010, 132, 7273-7275, DOI: 10.1021/ja9107885
(4) Schreiner, P. R.; Reisenauer, H. P.; Ley, D.; Gerbig, D.; Wu, C.-H.; Allen, W. D., "Methylhydroxycarbene: Tunneling Control of a Chemical Reaction," Science, 2011, 332, 1300-1303, DOI: 10.1126/science.1203761.
(5) Ley, D.; Gerbig, D.; Wagner, J. P.; Reisenauer, H. P.; Schreiner, P. R., "Cyclopropylhydroxycarbene," J. Am. Chem. Soc., 2011, 133, 13614-13621, DOI: 10.1021/ja204507j