Somehow I missed this paper when it came out a few months ago, even though I was aware it was coming – as I mentioned it in one of my previous posts!
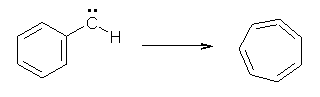
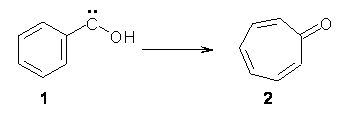
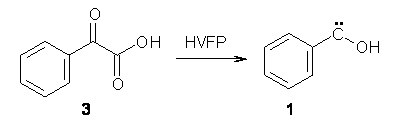
Anyways, Schreiner and Allen reported on their third study of hydroxyl carbenes (see these posts on dihydroxymethylene and hydroxymethylene), this time examining phenylhydroxycarbene.1 As I covered in my book, there is a lot of work on phenylcarbenes which typically ring expand to the cycloheptatetraene, see Reaction 1. One might expect phenylhydroxycarbene to do the same thing, i.e. 1 converting into 2 (Reaction 2). 1 is prepared by high-vacuum flash pyrolysis of phenylglyoxylic acid 3 and then capturing the product in an argon matrix at 11 K (Reaction 3).
|
|
Reaction 1 |
|
|
Reaction 2 |
|
|
Reaction 3 |
The carbene 1 is identified through comparison of its experimental and computed (anharmonic frequencies at CCSD(T)/cc-pVDZ) IR frequencies.
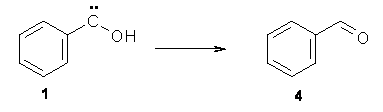
No ring expansion is observed at all – Reaction 2 does not occur. Instead, 1 rearranges to benzaldehyde 4 (Reaction 4) at 11 K with a half life of 2.46 h (and a half life of 2.55 h at 20 K). The deuterated analogue does not convert to benzaldehyde and 1-d appears to be completely stable.
|
|
Reaction 4 |
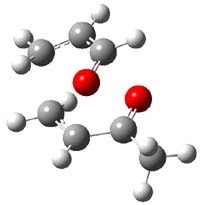
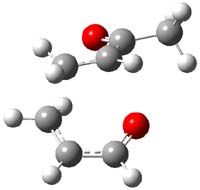
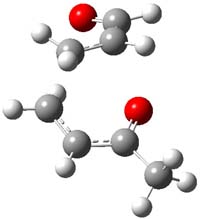
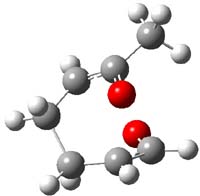
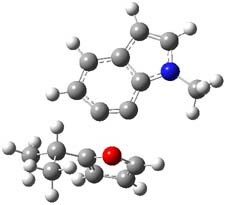
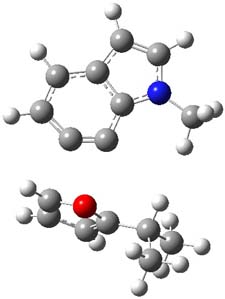
So, what is going on? The cis and trans forms of 1 interconvert through a barrier of 22.7 kcal mol-1. The trans isomer can convert to benzaldehyde (the reaction is very exothermic: -50.8 kcal mol-1) with a barrier of 28.8 kcal mol-1 through TS1, shown in Figure 1. The cis isomer can cleave into benzene and CO (not observed) with a huge barrier of 55 kcal mol-1. All of these barrier were computed at CCSD(T)/cc-pVQZ.
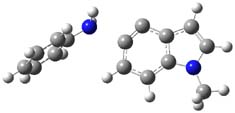
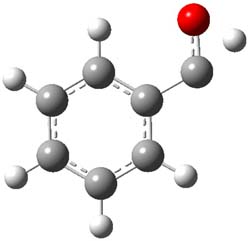 TS1 |
Figure 1. MP2/cc-pVDZ optimized transition state for the conversion of 1 into 4.
Benzaldehyde seems to be produced by passing through a huge barrier, something that is impossible from a thermal perspective (we’re at 11 K!). But this can be accomplished by tunneling. Tunneling probabilities were computed from the MP2/aug-cc-pVDZ intrinsic reaction path with barrier penetration integrals computed with the WKB approximation. The bottom line: the computed half-life is 3.3 h and the deuterated species is computed to have a half-life of 8700 years(!), both in excellent agreement with experimental observation. Quantum mechanical tunneling is clearly the explanation for this chemistry.
This is another fine example of the power of joint experimental/computational studies. And be on the look-out for an even more exciting case from this group. I met with Wes Allen on my recent trip to the University of Georgia and was entertained with another hydroxycarbene that undergoes quite novel tunneling!
References
(1) Gerbig, D.; Reisenauer, H. P.; Wu, C.-H.; Ley, D.; Allen, W. D.; Schreiner, P. R., "Phenylhydroxycarbene," J. Am. Chem. Soc., 2010, 132, 7273-7275, DOI: 10.1021/ja9107885
InChIs
1: InChI=1/C7H6O/c8-6-7-4-2-1-3-5-7/h1-5,8H
InChIKey=QVZIGMRPQWIGCV-UHFFFAOYAE
2: InChI=1/C7H6O/c8-7-5-3-1-2-4-6-7/h1-6H
InChIKey=QVWDCTQRORVHHT-UHFFFAOYAM
3: InChI=1/C8H6O3/c9-7(8(10)11)6-4-2-1-3-5-6/h1-5H,(H,10,11)/f/h10H
InChIKey=FAQJJMHZNSSFSM-KZFATGLACS
4: InChI=1/C7H6O/c8-6-7-4-2-1-3-5-7/h1-6H
InChIKey=HUMNYLRZRPPJDN-UHFFFAOYAE