Interacting bis-allyl radicals are the topic of a computational study by Gleiter and Borden.1 The new twist is to have the two allyl groups interact through a cyclobutyl, cyclopentyl or cyclohexyl ring, as in 1-3.

The degree of interaction of the radical electrons is evaluated with a number of metrics. First, the singlet-triplet energy gap is computed at CASSCF(6,6)/6-31G(d) and UB3LYP/6-31G(d). A larger gap is suggestive of strong interaction between the two allyl radicals. Next, the <S2> value of the UB3LYP wavefunction will be 0 for a pure singlet, which occurs when the radicals are strongly interacting. A value near 1 suggests an electron localized into each allyl fragment. Lastly, the natural orbital occupation numbers (NOON) of the two highest lying orbitals would be 2 and 0 for the pure interacting state and each would be 1 for the non-interacting state. The B3LYP/6-31G(d) optimized geometries of 1-3 are shown in Figure 1. The values of each metric are listed in Table 1.
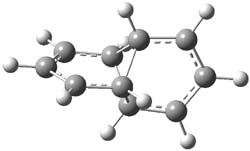 1 |
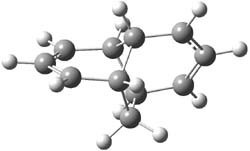 2 |
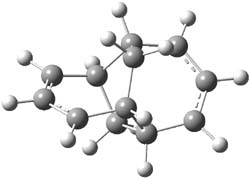 3 |
|
Figure 1. B3LYP/6-31G(d) optimized geometries of 1-3.
Table 1. Metrics for evaluating the allyl interaction in 1-3.
|
Diradical |
ΔEST (DFT)a |
ΔEST (CAS)a |
<S2> |
NOON |
|
1 |
21.4 |
25.5 |
0.0 |
1.62, 0.38 |
|
2 |
3.7 |
5.9 |
0.85 |
1.31, 0.69 |
|
3 |
1.6 |
2.4 |
0.96 |
1.20, 0.80 |
The different metrics are all consistent. The allyl radicals are strongly interacting in 1, with a low lying singlet state. The interaction is significantly lessened in 2 and smaller still in 3. The authors argue these differences in terms of the molecular orbital interactions between the allyl fragments and the central ring fragment.
References
(1) Lovitt, C. F.; Dong, H.; Hrovat, D. A.; Gleiter, R.; Borden, W. T., "Through-Bond Interactions in the Diradical Intermediates Formed in the Rearrangements of Bicyclo[n.m.0]alkatetraenes," J. Am. Chem. Soc., 2010, 132, 14617-14624, DOI: ja106329t
InChIs
1: InChI=1/C10H10/c1-3-7-9-5-2-6-10(7)8(9)4-1/h1-10H
InChIKey=QQZALYREQJSRLB-UHFFFAOYAA
2: InChI=1/C11H12/c1-3-8-7-9-4-2-6-11(8)10(9)5-1/h1-6,8-11H,7H2
InChIKey=XHSRXRHTBHVJQX-UHFFFAOYAV
3: InChI=1/C12H14/c1-3-9-7-12-6-2-5-11(9)8-10(12)4-1/h1-6,9-12H,7-8H2
InChIKey=JFNCWTOWDGQJLS-UHFFFAOYAA

Henry Rzepa responded on 07 Jan 2011 at 1:49 am #
This fascinating series of molecules reminds one of [1.1.1] propellane, where the terminal carbons might be represented, just as 1, as a coupled biradical .C(CH2)3C. As for 1, this propellane has a large singlet-triplet gap etc etc, and the spin-coupled end electrons are thought to create a (weak) charge-shift bond along the central axis of the molecule. That it probably is a bond is indicated by ρ(r) and the Laplacian at the bond critical point in the experimentally determined electron density for a propellane derivative.
So I note here that no QTAIM for 1 in particular is reported!
Henry Rzepa responded on 07 Jan 2011 at 2:27 am #
A postscript to previous comment. A QTAIM (B3LYP/6-311G(d,p) ) on 1 indicates three ring critical points lying along the D2d axis of the molecule, connecting if you will the two coupled biradical centres. The Laplacians at all three, and particularly the central one, are prominently positive (+0.3). I am not sure if one can say that a bond, by any normal definition connects them, but if it did, it would probably be a charge-shift type.