The click reaction has become a major workhorse of synthetic chemists since its proposal in 2001.1 Despite its efficiencies, no clear-cut example of its use in nature has been reported until 2012, where Yu and co-workers speculated that it might be utilized in the biosynthesis of lycojaponicumin A and B.2 Krenske, Patel, and Houk have examined the possibility of an enzyme activated click process in forming this natural product.3
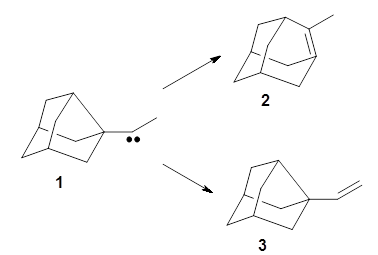
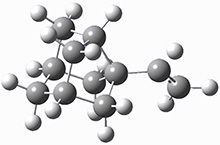
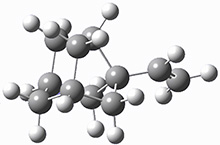
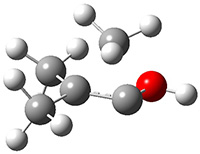
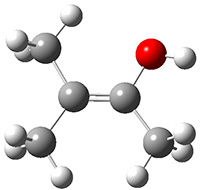
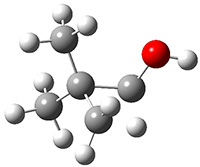
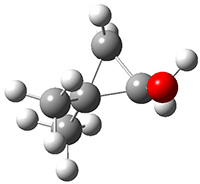
First they examined the gas-phase intramolecular [3+2] reaction that takes 1 into 2.
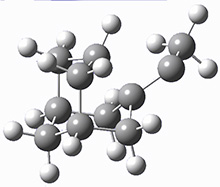
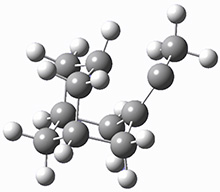
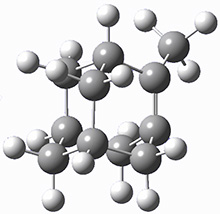
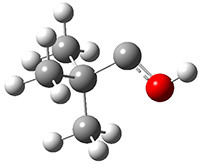
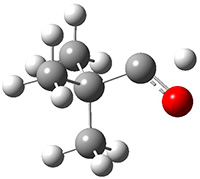
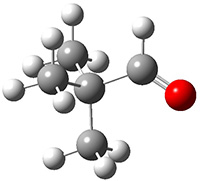
They identified (at M06-2X/def2-TZVPP/M06-2X/6-31+G(d,p)) four different low-energy conformations of 1, of which three have the proper orientation for the cyclization to occur. The lowest energy conformer, the TS, and the product 2 are shown in Figure 1. The free energy activation barrier in the gas phase is 19.8 kcal mol-1. Inclusion of water as an implicit solvent (through a TS starting from a different initial conformation) increases the barrier to 20.0 kcal mol-1. Inclusion of four explicit water molecules, hydrogen bonded to the nitrone and enone, predicts a barrier of 20.5 kcal mol-1. These values predict a slow reaction, but not totally impossible. In fact, Tantillo in a closely related work reported a theoretical study of the possibility of a [3+2] cyclization in the natural synthesis of flueggine A and virosaine, and found barriers of comparable size as here. Tantillo concludes that enzymatic activation is not essential.4
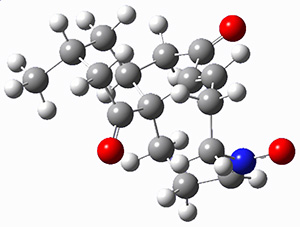 1 |
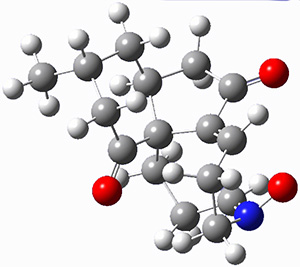 TS12 |
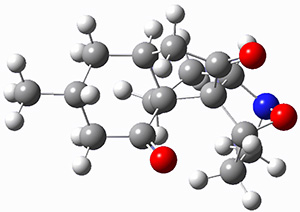 3 |
Table 1. M06-2X/6-31+G(d,p) optimized geometries of 1, TS12, and 2.
To model a potential enzyme, the Houk group created a theozyme whereby two water molecules act as hydrogen bond donors to the enone and the use of implicit solvent (diethyl ether) to mimic the interior of an enzyme. This theozyme model predicts a barrier of 15.3 kcal mol-1, or a 2000 fold acceleration of the click reaction. The search for such an enzyme might prove quite intriguing.
References
(1) Kolb, H. C.; Finn, M. G.; Sharpless, K. B. "Click Chemistry: Diverse Chemical Function from a Few Good Reactions," Angew. Chem. Int. Ed. 2001, 40, 2004-2021, DOI: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5.
(2) Wang, X.-J.; Zhang, G.-J.; Zhuang, P.-Y.; Zhang, Y.; Yu, S.-S.; Bao, X.-Q.; Zhang, D.; Yuan, Y.-H.; Chen, N.-H.; Ma, S.-g.; Qu, J.; Li, Y. "Lycojaponicumins A–C, Three Alkaloids with an Unprecedented Skeleton from Lycopodium japonicum," Org. Lett. 2012, 14, 2614-2617, DOI: 10.1021/ol3009478.
(3) Krenske, E. H.; Patel, A.; Houk, K. N. "Does Nature Click? Theoretical Prediction of an Enzyme-Catalyzed Transannular 1,3-Dipolar Cycloaddition in the Biosynthesis of Lycojaponicumins A and B," J. Am. Chem. Soc. 2013, 135, 17638-17642, DOI: 10.1021/ja409928z.
(4) Painter, P. P.; Pemberton, R. P.; Wong, B. M.; Ho, K. C.; Tantillo, D. J. "The Viability of Nitrone–Alkene (3 + 2) Cycloadditions in Alkaloid Biosynthesis," J. Org. Chem. 2014, 79, 432–435, DOI: 10.1021/jo402487d.
InChIs
1: InChI=1S/C16H21NO3/c1-11-8-12-10-14(18)13-4-2-6-17(20)7-3-5-16(12,13)15(19)9-11/h4,7,11-12H,2-3,5-6,8-10H2,1H3/b13-4-,17-7+
InChIKey=GVEYMXKHRRHCLV-KYGYAMEJSA-N
2: InChI=1S/C16H21NO3/c1-9-6-10-8-13(19)16-11(17-5-3-14(16)20-17)2-4-15(10,16)12(18)7-9/h9-11,14H,2-8H2,1H3/t9?,10-,11?,14?,15+,16-/m0/s1
InChIKey=QKFAOJHYKPBTKX-SGVNFLFUSA-N