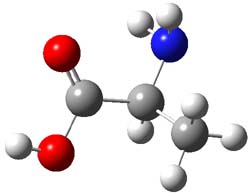
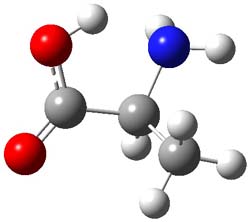
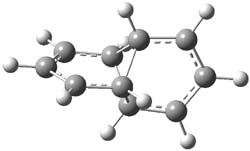
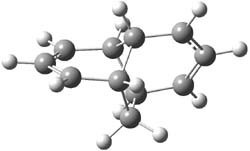
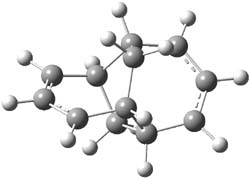
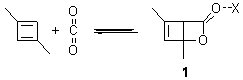
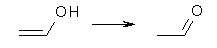Henry Rzepa’s response1 to the reported detection and x-ray structure of 1,3-dimethylcyclobutadiene2 has now been published. He takes a different tack than those take by Alabugin3 and Scheschkewitz4 in refuting the analysis of this work (see this earlier post). Rzepa discuses computations to evaluate the possible lifetime of 1,3-dimethylcyclobutadiene in the vicinity of CO2. In particular, he examines the barrier for the allowed [4+2] cycloaddition to give back the lactone 1 (Reaction 1), which was photolyzed in the experiment to produce the cyclobutadiene and CO2 species in the first place.
|
|
Reaction 1
|
The gas phase free energy barrier at 175 K (the experimental condition) computed at ωB97XD/6-311G(d,p) is 16.8 kcal mol-1, which is sufficiently high to limit this back reaction. Embedding this into a water continuum lowers the barrier to 12.9 kcal mol-1.
But the experiment has these species embedded inside a calixarene host along with guanidinum
cations. The cation could associate with the CO2 (indicated in Reaction 1 as X), and inclusion of a guanidinium in the gas phase, reduces the barrier to 3.3 kcal mol-1. Rerunning this computation now with a water continuum produce an intermediate zwitterion formed by making the C-C bond, and the second step makes the C-O bond.
Finally, modeling the reaction with guanidium inside a calixarene host leads to a barrier of 8 kcal
mol-1, 10.5 kcal mol-1 with water continuum. Rzepa concludes that recombination of 1,3-dimethylcyclobutadiene and CO2 to give 1 should be too fast on the timescale of the experiment for observation of the cyclobutadiene. This argument, along with the two previous papers, strongly casts doubt on the original claim.
I should point out that Henry has deposited all the structures in a nice enhanced table. You may need a subscription to get to this – I have not checked the access conditions.
References
(1) Rzepa, H. S., "Can 1,3-dimethylcyclobutadiene and carbon dioxide co-exist inside
a supramolecular cavity?," Chem. Commun. 2011, ASAP, DOI: 10.1039/C0CC04023A
(2) Legrand, Y.-M.; van der Lee, A.; Barboiu, M., "Single-Crystal X-ray Structure of 1,3-Dimethylcyclobutadiene by Confinement in a Crystalline Matrix," Science 2010, 329, 299-302, DOI: 10.1126/science.1188002.
(3) Alabugin, I. V.; Gold, B.; Shatruk, M.; Kovnir, K., "Comment on "Single-Crystal X-ray Structure of 1,3-Dimethylcyclobutadiene by Confinement in a Crystalline Matrix"," Science, 2010, 330, 1047, DOI: 10.1126/science.1196188.
(4) Scheschkewitz, D., "Comment on "Single-Crystal X-ray Structure of 1,3-Dimethylcyclobutadiene by Confinement in a Crystalline Matrix"," Science, 2010, 330, 1047, DOI: 10.1126/science.1195752.
InChIs
1: InChI=1/C7H8O2/c1-4-3-7(2)5(4)6(8)9-7/h3,5H,1-2H3
InChIKey=GLYAMHMFKKLRAL-UHFFFAOYAT
1,3-dimethylcyclobutadiene: InChI=1/C6H8/c1-5-3-6(2)4-5/h3-4H,1-2H3
InChIKey=ADQGKIKNUMJFSL-UHFFFAOYAU