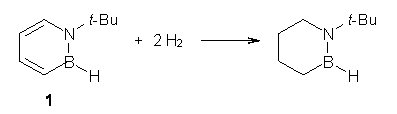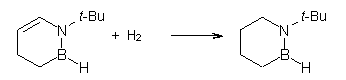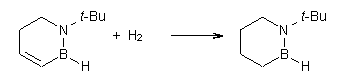Kinetic isotope effects (KIE) are a valuable tool for probing mechanisms without changing the potential energy surface. Their interpretation can sometimes be difficult – for example is a perdeutero group larger or smaller than the perhydro analogue?
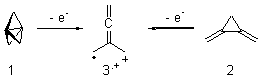
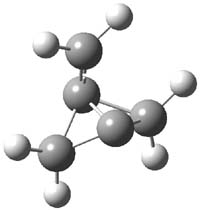
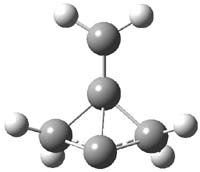
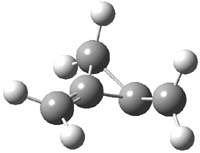
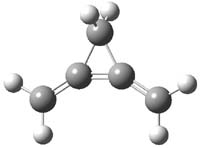
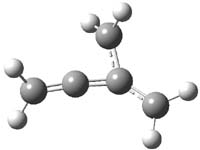
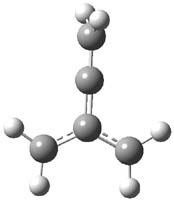
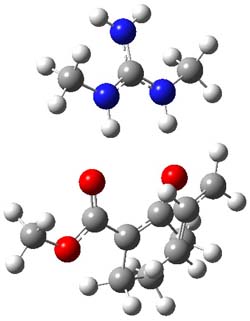
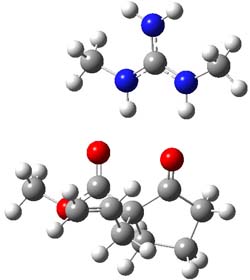
O’Leary, Rablen and Meyer have examined two related molecules and their KIEs relating to stereoinversion.1 1 exhibits a normal isotope effect (kH/kD = 1.06) while 2 has an inverse isotope effect (kH/kD = 0.880). They optimized the structures and transition states (see Figure 1) for racemization of both compounds at B3LYP and MP2, and computed isotope effects based on the Biegeleisen-Mayer equation (which is based on reduced partition functions). The KIEs obtained from the two computational methods is very similar.
|
|
|
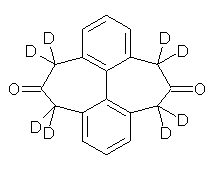
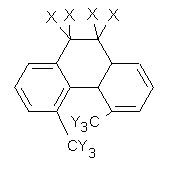
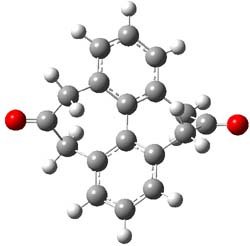 1 |
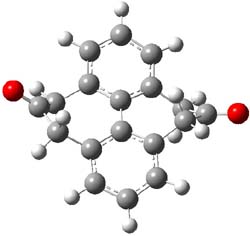 1TS |
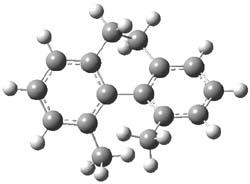 2 |
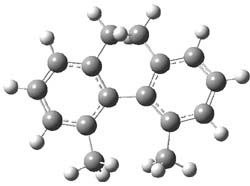 2TS |
Figure 1. MP2/6-31G(d,p) optimized geometries of 1 and 2 and the transition states for their racemization.
The experimental and computed KIEs are listed in Table 1. The agreement between experiment and computation is excellent – suggesting that computations should be routinely employed when analyzing isotope effects.
Table 1. Experimental and computed KIEs for racemization of 1 and 2.
|
|
Expt |
Comp |
|
d8–1 |
1.06 |
1.075 |
|
d6–2 |
0.880 |
0.888 |
|
d4–2 |
0.952 |
0.953 |
|
d10–2 |
0.847 |
0.846 |
The authors decompose the isotope effects into enthalpic and entropic components and note that the interplay between these two can be subtle – sometimes one might dominate and other times the second term will dominate, and the terms can be cooperative or non-cooperative.
References
(1) O’Leary, D. J.; Rablen, P. R.; Meyer, M. P., "On the Origin of Conformational Kinetic Isotope Effects," Angew. Chem. Int. Ed., 2011, 50, 2564-2567, DOI: 10.1002/anie.201007322
InChIs
1: InChI=1/C18H14O2/c19-15-7-11-3-1-4-12-8-16(20)10-14-6-2-5-13(9-15)18(14)17(11)12/h1-6H,7-10H2
InChIKey=DYZSIUYFWKNLHS-UHFFFAOYAB
d8–1: InChI=1/C18H14O2/c19-15-7-11-3-1-4-12-8-16(20)10-14-6-2-5-13(9-15)18(14)17(11)12/h1-6H,7-10H2/i7D2,8D2,9D2,10D2
InChIKey=DYZSIUYFWKNLHS-UFBJYANTEO
2: InChI=1/C16H18/c1-11-5-3-7-13-9-10-14-8-4-6-12(2)16(14)15(11)13/h3-8,13,15H,9-10H2,1-2H3
InChIKey=OBRIKDRTDGHGIQ-UHFFFAOYAU