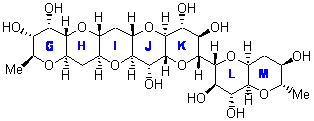Another three applications of computed NMR chemical shifts towards structure identification have appeared, dealing with carbohydrates and natural products.
Prediction of NMR Signals of Carbohydrates
The study by Cramer and Hoye1 investigates identification of diastereomers with NMR, in particular, identification of cis and trans isomers of 2-methyl- (1), 3-methyl- (2), and 4-methylcyclohexanol (3). The study discusses the ability of different DFT methods to predict the chemical shifts of these alcohols in regard to distinguishing their different configurations. An interesting twist is that they have developed a functional specifically suited to predict proton chemical shifts and a second functional specifically for predicting carbon chemical shifts.2

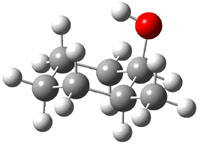
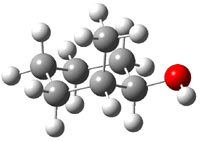
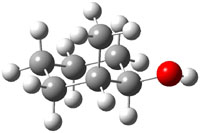
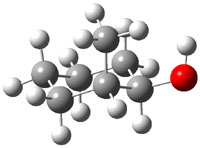
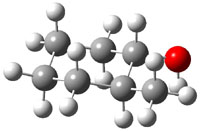
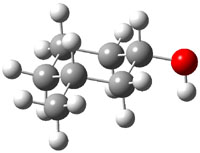
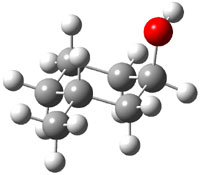
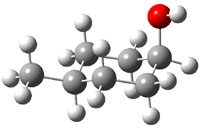
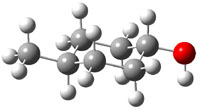
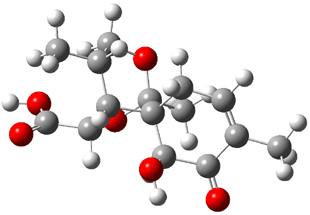
The approach they take was first to optimize the six different conformations for each diastereomer including solvent (chloroform). They chose to optimize the structures at B3LYP/6-311+G(2d,p) with PCM. The six conformers (notice the axial/equatorial relationships, along with the position of the alcohol hydrogen) of 1c are presented in Figure 1. Chemical shifts were then obtained with a number of different methods, weighting them according to a Boltzmann distribution.
Figure 1. PCM/B3LYP/6-311+G(2d,p) optimized structures of the conformers of 1c. Relative energies (kcal mol-1) are listed for each isomer.
Now a brief digression into how they developed their modified functional.2 They define the exchange-correlation functional (see Chapter 1.3.1 of my book – or many other computational chemistry books!) as
Exc = P2Ex(HF) + P3ΔEx(B) + P4Ex(LSDA) + P5ΔEc(LYP) + P6Ec(LSDA)
where the Ps are parameters to be fit and Ex(HF) is the Hartree-Fock exchange energy, ΔEx(B) is the Becke gradient correction to the local spin-density approximation (LSDA), Ex(LSDA) is the exchange energy, ΔEc(LYP) is the Lee-Yang-Parr correction to the LSDA correlation energy, and Ec(LSDA) is the LSDA correlation energy. Chemical shifts were computed for proton and carbon, and the parameters P were adjusted (between 0 and 1) to minimize the error in the predicted chemical shifts from the experimental values. A total of 43 different molecules were used for this fitting procedure. The values of the parameters are given for the carbon functional (WC04), the proton functional (WP04) and B3LYP (as a reference) in Table 1. Note that there is substantial difference in the values of the parameter among these three different functionals.
Table 1. Values of the parameters P for the functionals WC04, WP04, and B3LYP.
|
|
|
P2
|
P3
|
P4
|
P5
|
P6
|
|
|
WC04
|
0.7400
|
0.9999
|
0.0001
|
0.0001
|
0.9999
|
|
WP04
|
0.1189
|
0.9614
|
0.999
|
0.0001
|
0.9999
|
|
B3LYP
|
0.20
|
0.72
|
0.80
|
0.81
|
1.00
|
|
Now, the computed proton and carbon chemical shifts using 4 different functions (B3LYP, PBE1, MP04, and WC04) for 1-3 were compared with the experiment values. This comparison was made in a number of different ways, but perhaps most compellingly by looking at the correlation coefficient of the computed shifts compared with the experimental shifts. This was done for each diastereomer, i.e. the computed shifts for 2c and 2t were compared with the experimental shifts of both 2c and 2t. If the functional works well, the correlation between the computed and experimental chemical shifts of 2c (and 2t) should be near unity, while the correlation between the computed shifts of 2c and the experimental shifts of 2t should be dramatically smaller than one. This is in fact the case for all three functionals. The results are shown in Table 2 for B3LYP and WP04, with the later performing slightly better. The results for the carbon shifts are less satisfactory; the correlation coefficients are roughly the same for all comparisons with B3LYP and PBE1, and WC04 is only slightly improved.
Nonetheless, the study clearly demonstrates the ability of DFT-computed proton chemical shifts to discriminate between diasteromers.
Table 2. Correlation coefficients between the computed and experimental proton chemical shifts.a
|
|
|
2ccomp
(1.06)
xyz
|
2tcomp
(0.0)
xyz
|
|
|
2cexp
2texp
|
0.9971
0.9985
0.8167
0.8098
|
0.8334
0.9050
0.9957
0.9843
|
|
|
|
3c
(0.0)
xyz
|
3t
(0.63)
xyz
|
|
|
3cexp
3texp
|
0.9950
0.9899
0.8856
0.9310
|
0.8763
0.8717
0.9990
0.9979
|
|
|
|
4c
(0.54)
xyz
|
4t
(0.0)
xyz
|
|
|
4cexp
4texp
|
0.9993
0.9975
0.8744
0.8675
|
0.8335
0.9279
0.9983
0.9938
|
|
aPCM/B3LYP/6-311+G(2d,p)//PCM/ B3LYP/6-31G(d) in regular type and PCM/WP04/6-311+G(2d,p)//PCM/ B3LYP/6-31G(d) in italic type. Relative energy (kcal mol-1) of the most favorable conformer of each diastereomer is given in parenthesis.
Predicting NMR of Natural Products
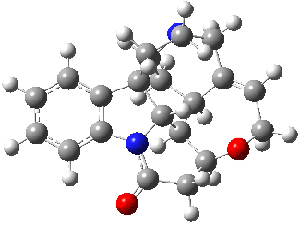
Bagno has a long-standing interest in ab initio prediction of NMR. In a recent article, his group takes on the prediction of a number of complex natural products.3 As a benchmark, they first calculated the NMR spectra of strychnine (4) and compare it with its experimental spectrum. The optimized PBE1PBE/6-31G(d,p) geometry of 4 is drawn in Figure 2. The correlation between the computed NMR chemical shifts for both 1H and 13C is quite good, as seen in Table 3. The corrected mean average errors are all very small, but Bagno does point out that four pairs of proton chemical shifts and three pairs of carbon chemical shifts are misordered.
|
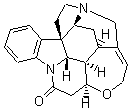
Strychnine
4
|
|
Figure 2. PBE1PBE/6-31G(d,p) geometry of strychnine 4.3
Table 3. Correlation coefficient and corrected mean average error
(CMAE) between the computed and experiment chemical shifts of 4.
|
|
|
δ(1H)
|
δ(13C)
|
|
method
|
r2
|
CMAE
|
r2
|
CMAE
|
|
B3LYP/cc-pVTZ
|
0.9977
|
0.07
|
0.9979
|
1.4
|
|
PBE1PBE/cc-pVTZ
|
0.9974
|
0.08
|
0.9985
|
0.9
|
|
The study of the sesquiterpene carianlactone (5) demonstrates the importance of including solvent in the NMR computation. The optimized B3LYP/6-31G(d,p) geometry of 5 is shown in Figure 3, and the results of the comparison of the computed and experimental chemical are listed in Table 4. The correlation coefficient is unacceptable when the x-ray structure is used. The agreement improves when the gas phase optimized geometry is employed, but the coefficient is still too far from unity. However, optimization using PCM (with the solvent as pyridine to match experiments) and then computing the NMR chemical shifts in this reaction field provides quite acceptable agreement between the computed and experimental chemical shifts.
|
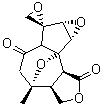
Corianlactone 5
|
|
Figure 3. B3LYP/6-31G(d,p) geometry of carianlactone 5.3
Table 4. Correlation coefficient and corrected mean average error (CMAE) between
the computed and experiment chemical shifts of 5.
|
|
|
δ(1H)
|
δ(13C)
|
|
geometry
|
r2
|
CMAE
|
r2
|
CMAE
|
|
X-ray
|
0.9268
|
0.23
|
0.9942
|
3.1
|
|
B3LYP/6-31G(d,p)
|
0.9513
|
0.19
|
0.9985
|
1.6
|
|
B3LYP/6-31G(d,p) + PCM
|
0.9805
|
0.11
|
0.9990
|
1.2
|
|
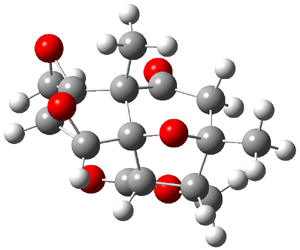
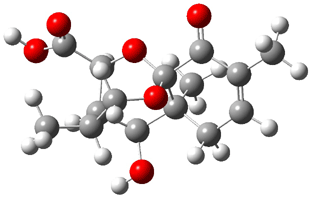
Lastly, Bagno took on the challenging structure of the natural product first identified as boletunone B (6a).4 Shortly thereafter, Steglich reinterpreted the spectrum and gave the compound the name isocyclocalopin A (6b).5 A key component of the revised structure was based on the δ 0.97 ppm signal that they assigned to a methyl above the enone group, noting that no methyl in 6a should have such a high field shift.
Bagno optimized the structures of 6a and 6b at B3LYP/6-31G(d,p), shown in Figure 4. The NMR spectra for 6a and 6b were computed with PCM (modeling DMSO as the solvent). The correlation coefficients and CMAE are much better for the 6b model than for the 6a model., supporting the reassigned structure. However, the computed chemical shift for the protons of the key methyl group in question are nearly identical in the two proposed structures: 1.08 ppm in 6a and 1.02 ppm in 6b. Nonetheless, the computed chemical shifts and coupling constants of 6b are a better fit with the experiment than those of 6a.
|
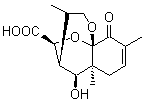
boletunone B 6a
|
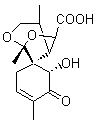
isocyclocalopin A 6b
|
|
|
|
Figure 4. B3LYP/6-31G(d,p) geometry of the proposed structures of Boletunone B, 6a and 6b.3
Table 5. Correlation coefficient and corrected mean average error (CMAE) between the computed (B3LYP/6-31G(d,p) + PCM) and experiment chemical shifts of 6a and 6b.
|
|
|
δ(1H)
|
δ(13C)
|
|
structure
|
r2
|
CMAE
|
r2
|
CMAE
|
|
6a
|
0.9675
|
0.22
|
0.9952
|
3.7
|
|
6b
|
0.9844
|
0.15
|
0.9984
|
1.9
|
|
In a similar vein, Nicolaou and Frederick has examined the somewhat controversial structure of maitotxin.6 For the sake of brevity, I will not draw out the structure of maitotxin; the interested reader should check out its entry in wikipedia. The structure of maitotoxin has been extensively studied, but in 2006, Gallimore and Spencer7 questioned the stereochemistry of the J/K ring juncture. A fragment of maitotoxin that has the previously proposed stetreochemistry is 7. Gallimore and Spencer argued for a reversed stereochemistry at this juncture (8), one that would be more consistent with the biochemical synthesis of the maitotoxin. Nicolaou noted that reversing this stereochemistry would lead to other stereochemical changes in order for the structure to be consistent with the NMR spectrum. Their alternative is given as 9.
Nicolaou and Freferick computed 13C NMR of the three proposed fragments 7-9 at B3LYP/6-31G*; unfortunately they do not provide the coordinates. They benchmark this method against brevetoxin B, where the average error is 1.24 ppm, but they provide no error analysis – particularly no regression so that corrected chemical shift data might be employed. The best agreement between the computed and experimental chemical shifts is for 7, with average difference of 2.01 ppm. The differences are 2.85 ppm for 8 and 2.42 ppm for 9. These computations support the original structure of maitotoxin. The Curious Wavefunction blog discusses this topic, with an emphasis on the possible biochemical implication.
References
(1) Wiitala, K. W.; Al-Rashid, Z. F.; Dvornikovs, V.; Hoye, T. R.; Cramer, C. J., "Evaluation of Various DFT Protocols for Computing 1H and 13C Chemical Shifts to Distinguish Stereoisomers: Diastereomeric 2-, 3-, and 4-Methylcyclohexanols as a Test Set," J. Phys. Org. Chem. 2007, 20, 345-354, DOI: 10.1002/poc.1151
(2) Wiitala, K. W.; Hoye, T. R.; Cramer, C. J., "Hybrid Density Functional Methods Empirically Optimized for the Computation of 13C and 1H Chemical Shifts in Chloroform Solution," J. Chem. Theory Comput. 2006, 2, 1085-1092, DOI: 10.1021/ct6001016
(3) Bagno, A.; Rastrelli, F.; Saielli, G., "Toward the Complete Prediction of the 1H and 13C NMR Spectra of Complex Organic Molecules by DFT Methods: Application to Natural Substances," Chem. Eur. J. 2006, 12, 5514-5525, DOI: 10.1002/chem.200501583
(4) Kim, W. G.; Kim, J. W.; Ryoo, I. J.; Kim, J. P.; Kim, Y. H.; Yoo, I. D., "Boletunones A and B, Highly Functionalized Novel Sesquiterpenes from Boletus calopus," Org. Lett. 2004, 6, 823-826, DOI: 10.1021/ol049953i
(5) Steglich, W.; Hellwig, V., "Revision of the Structures Assigned to the Fungal Metabolites Boletunones A and B," Org. Lett. 2004, 6, 3175-3177, DOI: 10.1021/ol048724t.
(6) Nicolaou, K. C.; Frederick, M. O., "On the Structure of Maitotoxin," Angew. Chem. Int. Ed., 2007, 46, 5278-5282, DOI: 10.1002/anie.200604656.
(7) Gallimore, A. R.; Spencer, J. B., "Stereochemical Uniformity in Marine Polyether Ladders – Implications for the Biosynthesis and Structure of Maitotoxin," Angew. Chem. Int. Ed. 2006, 45, 4406-4413, DOI: 10.1002/anie.200504284.
InChI
1: InChI=1/C7H14O/c1-6-4-2-3-5-7(6)8/h6-8H,2-5H2,1H3
2: InChI=1/C7H14O/c1-6-3-2-4-7(8)5-6/h6-8H,2-5H2,1H3
3: InChI=1/C7H14O/c1-6-2-4-7(8)5-3-6/h6-8H,2-5H2,1H3
4: InChI=1/C21H22N2O2/c24-18-10-16-19-13-9-17-21(6-7-22(17)11-12(13)5-8-25-16)14-3-1-2-4-15(14)23(18)20(19)21/h1-5,13,16-17,19-20H,6-11H
5: InChI=1/C14H14O6/c1-12-2-6(15)8-13(4-18-13)9-10(19-9)14(8,20-12)7-5(12)3-17-11(7)16/h5,7-10H,2-4H2,1H3/t5-,7-,8?,9+,10+,12+,13?,14-/m1/s1
6a: InChI=1/C15H20O6/c1-7-4-5-14(3)12(17)9-8(2)6-20-15(14,11(7)16)21-10(9)13(18)19/h4,8-10,12,17H,5-6H2,1-3H3,(H,18,19)/t8-,9+,10+,12+,14-,15-/m1/s1
6b: InChI=1/C15H20O6/c1-7-4-5-15(12(17)10(7)16)9-8(2)6-20-14(15,3)21-11(9)13(18)19/h4,8-9,11-12,17H,5-6H2,1-3H3,(H,18,19)/t8?,9-,11?,12+,14-,15-/m1/s1