An emerging theme in this blog is Möbius systems, ones that can be aromatic or antiaromatic. Rzepa has led the way here, especially in examining annulenes with a twisted structure. Along with Schleyer and Schaefer, they have now explored a series of Möbius annulenes.1 The particularly novel aspect of this new work is the examination of higher-order Möbius systems. In the commonly held notion of the Möbius strip, the strip contains a single half twist. Rzepa points out that the notion of twist must be considered as two parts, a part due to torsions and a part due to writhe.2 We can think of the Möbius strip as formed by a ladder where the ends are connect such that the left bottom post connects with the top right post and the bottom right post connects with the top left post. Let’s now consider the circle created by joining the midpoints of each rug of the ladder. If this circle lies in a plane, then the torsion is π/N where N is the number of rungs in the ladder. But, the collection of midpoints does not have to lie in a plane, and if these points distort out of plane, that’s writhe and allows for less torsion in the strip.The sum of these two parts is called Lk and it will be an integral multiple of π. So the common Möbius strip has Lk = 1.
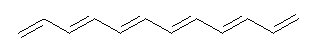
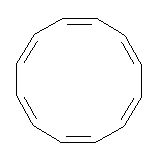
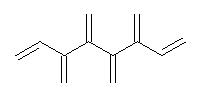
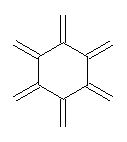
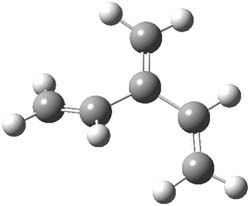
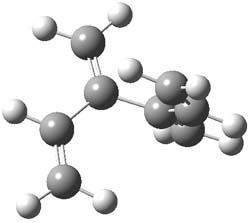
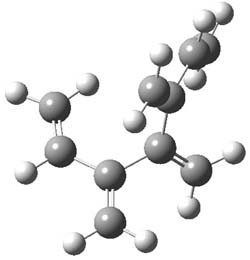
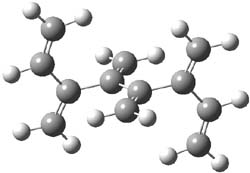
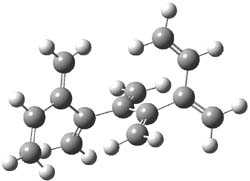
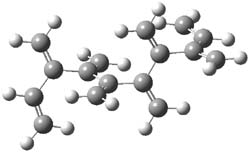
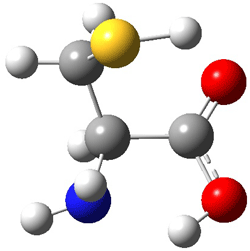
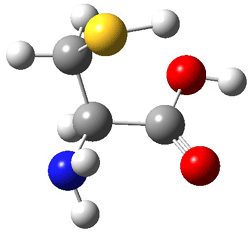
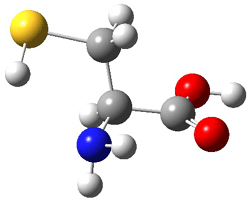
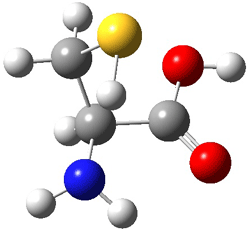
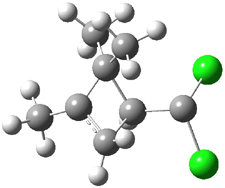
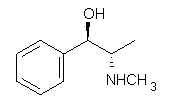
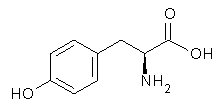
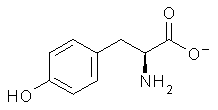
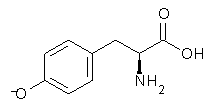
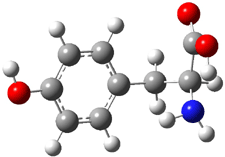
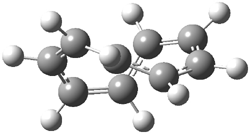
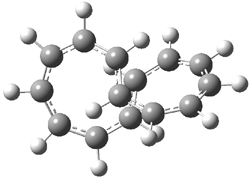
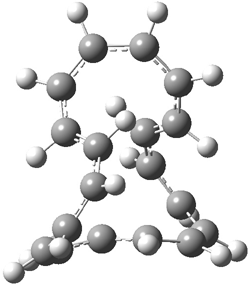
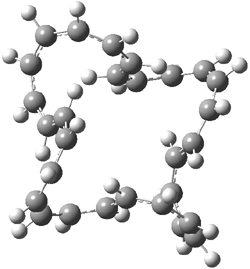
An example of a molecular analogue of the common Möbius strip is the annulene C9H9+ (1) – see figure 1. But Möbius strips can have more than one twist. Rzepa, Schleyer, and Schaefer have found examples with Lk = 2, 3, or 4. Examples are C14H14 (2) with one full twist (Lk = 2, two half twists), C16H162- (3) with three half twists, and C20H202+ (4) with four half twists.
 1 |
 2 |
 3 |
 4 |
Figure 1. Structures of annulenes 1-4.
These annulenes with higher-order twisting, namely 2-4, are aromatic, as determined by a variety of measures. For example, all express negative NICS values, all have positive diagmagnetic exaltations, and all express positive isomerization stabilization energies (which are a measure of aromatic stabilization energy).
References
(1) Wannere, C. S.; Rzepa, H. S.; Rinderspacher, B. C.; Paul, A.; Allan, C. S. M.; Schaefer Iii, H. F.; Schleyer, P. v. R., "The Geometry and Electronic Topology of Higher-Order Charged M&oml;bius Annulenes" J. Phys. Chem. A 2009, ASAP, DOI: 10.1021/jp902176a
(2) Fowler, P. W.; Rzepa, H. S., "Aromaticity rules for cycles with arbitrary numbers of half-twists," Phys. Chem. Chem. Phys. 2006, 8, 1775-1777, DOI: 10.1039/b601655c.