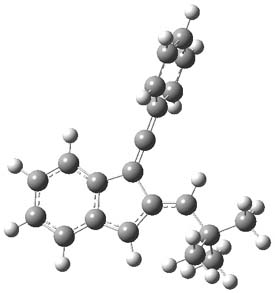
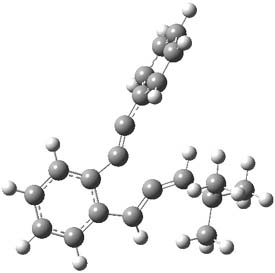
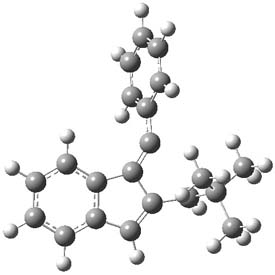
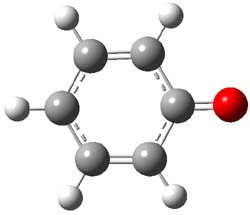
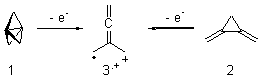
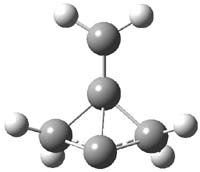
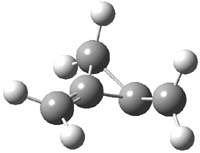
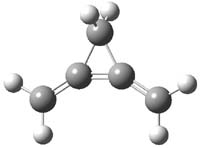
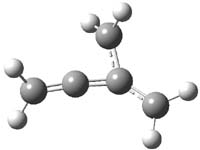
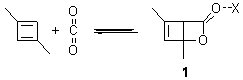
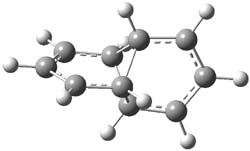
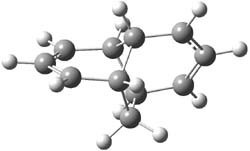
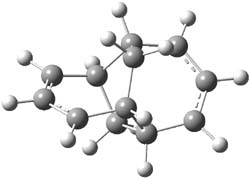
Earlier this year, Barboiu made the astonishing claim of the x-ray characterization of 1,3-dimethylcyclobutadiene, brought about by the photolysis of 4,6-dimethyl-α-pyrone encapsulated in a guanidinium-sulfonate-calixarene crystal (Reaction 1).1 I had not blogged on this paper because Henry Rzepa did a quite thorough analysis of it in this blog post. Now, a couple of rebuttals have appeared and it is time to examine this study.

Alabugin calls in question whether the reaction has in fact proceeded beyond 2.2 They note that in the x-ray crystal structure, the distance between a carbon of the purported cyclobutadiene ring and the carbon of CO2 is only 1.50 and 1.61 Å. Barboiu called this a “strong van der Waals contact”, but this is a distance much more attributable to a covalent bond. In fact, the shorter distance is in fact shorter than some of the other C-C distances in the structure that Barboiu calls covalent! Perhaps more bizarre is that the putative CO2 fragment is highly bent: 119.9&;deg;, a value inconsistent with CO2 but perfectly ordinary for an sp2 carbon. In fact, B3LYP/6-31G** computations suggest that bending CO2 this much costs about 75 kcal mol-1 – and tack on another 7 kcal mol-1 to make the two C-O distances unequal (as found in the x-ray structure!). Thus, Alabugin suggests that only 2 has been formed, and notes that the cleavage to 3 would likely require light of much higher energy that that used in the Barboiu experiment.
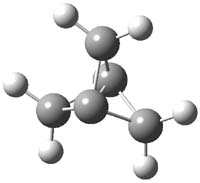
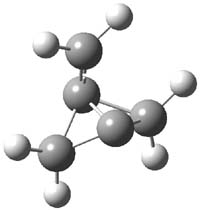
Scheschkewitz argues that the x-ray data can be better interpreted as suggesting only the Dewar β-lactone 2 is present, though in its two enantiomeric forms.3 There is no evidence, he suggests of any cyclobutadiene component at all.
It should be noted that Barboiu stands4 by his original work and original assignment, claiming that these types of x-ray experiments are quite difficult and large error bars in atom positions are inherent to the study.
Henry Rzepa has blogged again on this controversy and has a paper coming out on this soon. I shall update when it appears. Henry notes in one of the comments to his blog that a TD-DFT computations does show that the Dewar β-lactone 2 is transparent from 320-500nm.
References
(1) Legrand, Y.-M.; van der Lee, A.; Barboiu, M., "Single-Crystal X-ray Structure of 1,3-Dimethylcyclobutadiene by Confinement in a Crystalline Matrix," Science 2010, 329, 299-302, DOI: 10.1126/science.1188002.
(2) Alabugin, I. V.; Gold, B.; Shatruk, M.; Kovnir, K., "Comment on "Single-Crystal X-ray Structure of 1,3-Dimethylcyclobutadiene by Confinement in a Crystalline Matrix"," Science, 330, 1047, DOI: 10.1126/science.1196188.
(3) Scheschkewitz, D., "Comment on "Single-Crystal X-ray Structure of 1,3-Dimethylcyclobutadiene by Confinement in a Crystalline Matrix"," Science 2010, 330, 1047, DOI: 10.1126/science.1195752.
(4) Legrand, Y.-M.; van der Lee, A.; Barboiu, M., "Response to Comments on "Single-Crystal X-ray Structure of 1,3-Dimethylcyclobutadiene by Confinement in a Crystalline Matrix"," Science, 330, 1047, DOI: 10.1126/science.1195846.
InChIs
1: InChI=1/C7H8O2/c1-5-3-6(2)9-7(8)4-5/h3-4H,1-2H3
InChIKey=IXYLIUKQQQXXON-UHFFFAOYA
2: InChI=1/C7H8O2/c1-4-3-7(2)5(4)6(8)9-7/h3,5H,1-2H3
InChIKey=GLYAMHMFKKLRAL-UHFFFAOYAT
3: InChI=1/C6H8/c1-5-3-6(2)4-5/h3-4H,1-2H3
InChIKey=ADQGKIKNUMJFSL-UHFFFAOYAU