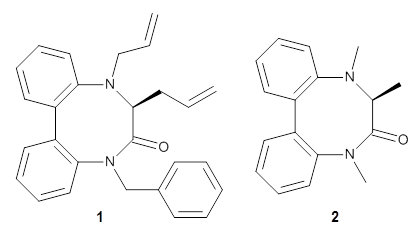
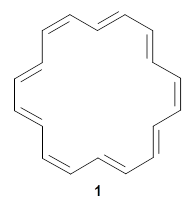
Bunz and co-workers have synthesized the novel aromatic compound 1 that contains 13 acenes in a row.1
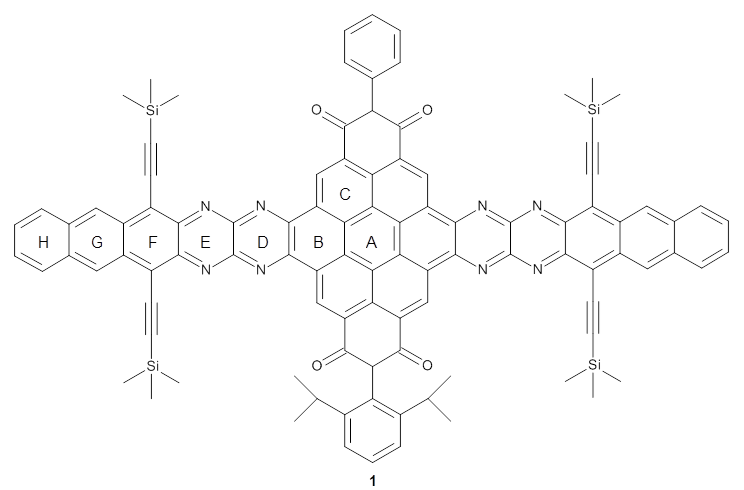
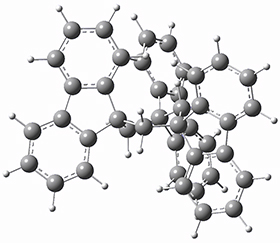
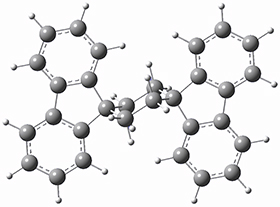
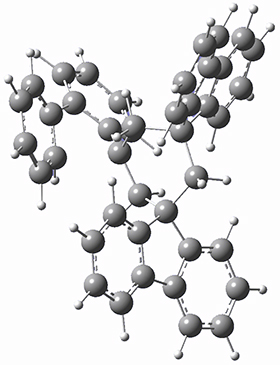
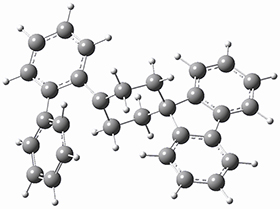
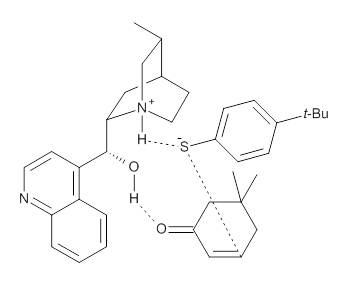
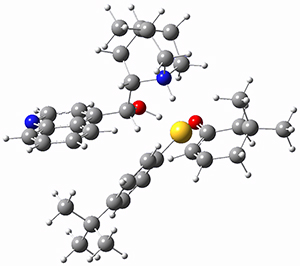
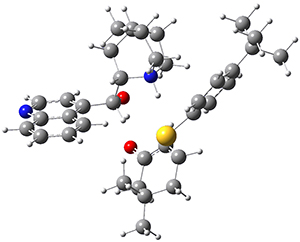
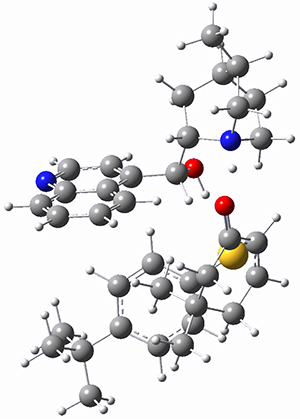
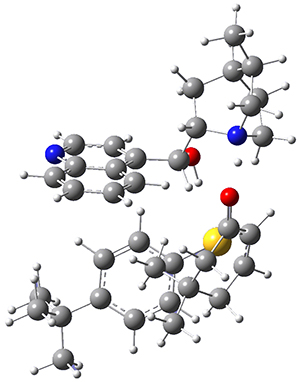
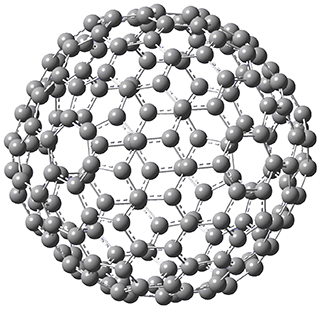
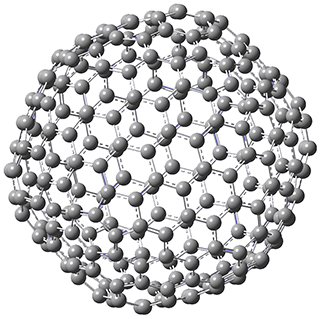
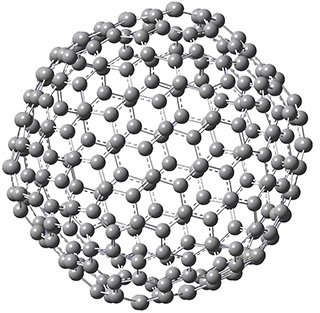
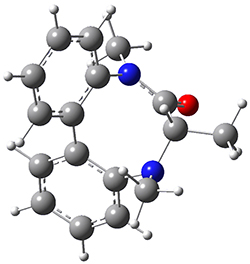
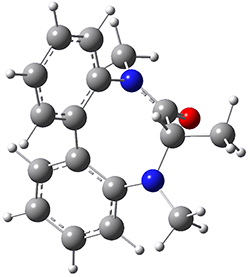
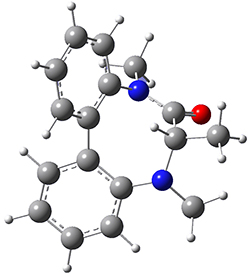
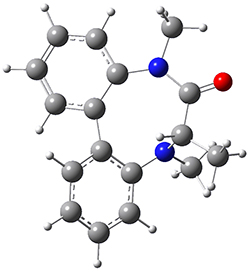
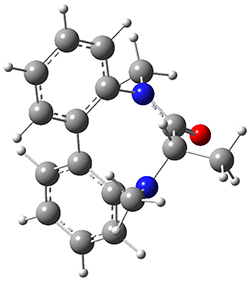
They optimized the geometry of 1 at B3LYP/6-311G*, and its geometry is shown in Figure 1. Even though this compound has quite an extensive π-system, an unrestricted computations collapses to the closed-shell wavefunction.
1 |
Figure 1. B3LYP/6-311G* optimized geometry of 1. (As always, don’t forget to click on this image to launch JMol and visualize the molecule in 3-D.)
NICS(1)πzz values for the rings are given in Table 1. Interestingly, the aromaticity of the coronene moiety is reduced; in fact the central ring (ring A, with rings labeled sequentially working towards either end from the center) has a very small NICS value of only -3.77.
Table 1. NICS(1)πzz values for the rings of 1.
|
Ring |
NICS(1)πzz |
|
A |
-3.7 |
References
(1) Endres, A. H.; Schaffroth, M.; Paulus, F.; Reiss, H.; Wadepohl, H.; Rominger, F.; Krämer, R.; Bunz, U. H. F. "Coronene-Containing N-Heteroarenes: 13 Rings in a Row," J. Am. Chem. Soc. 2016, 138, 1792-1795, DOI: 10.1021/jacs.5b12642.
InChIs
1: InChI=1S/C100H76N8O4Si4/c1-49(2)56-31-24-32-57(50(3)4)75(56)84-95(111)72-47-68-80-78-66(89-91(68)107-99-97(105-89)101-85-58(33-37-113(5,6)7)62-41-52-27-20-22-29-54(52)43-64(62)60(87(85)103-99)35-39-115(11,12)13)45-70-76-71(94(110)74(93(70)109)51-25-18-17-19-26-51)46-67-79(82(76)78)81-69(48-73(96(84)112)77(72)83(80)81)92-90(67)106-98-100(108-92)104-88-61(36-40-116(14,15)16)65-44-55-30-23-21-28-53(55)42-63(65)59(86(88)102-98)34-38-114(8,9)10/h17-32,41-50,74,84H,1-16H3
InChIKey=GNQHLGPUXJMSCH-UHFFFAOYSA-N