Structure determination has been greatly facilitated by the use of computed NMR spectra to compare with experimental spectra. Perhaps the best method for doing this is the DP4 procedure developed by Smith and Goodman.1 (I have a previous post on their paper.) The basic idea is that if you have an experimental NMR spectrum and a number of potential structures, the computed spectra for each possibility are ranked by a statistical treatment based on the Student t-test.
Grimblat, Zanardi, and Sarotti question a couple of the assumptions embedded within the DP4 method, and offer a revision that they call DP4+.2 The two assumptions are (1) that the chemical shifts are computed at B3LYP/6-31G**//MMFF and (2) that the chemical shifts are scaled and then utilized in the analysis.
To test these assumptions, they examine a set of 72 organic compounds comprising 1219 13C shifts and 1123 1H shifts. They optimized the structures at B3LYP/6-31G* and computed the chemical shifts of these compounds using the B3LYP and mPW1PW91 functionals with 6 basis sets (6-31G*, 6-31G**, 6-31+G**, 6-311G*, 6-311G**, and 6-311+G**). With all of the combinations, the standard deviation of both the proton and carbon chemical shifts were significantly smaller than with the originally proposed method.
With regards to the second assumption, they define a new probability functions that multiplies the error using scaled chemical shifts with the error using unscaled chemical shifts, and this they call DP4+. Again with all of the computational methods, the DP4+ prediction outperforms the DP4 prediction.
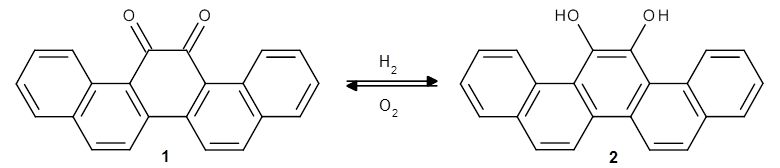
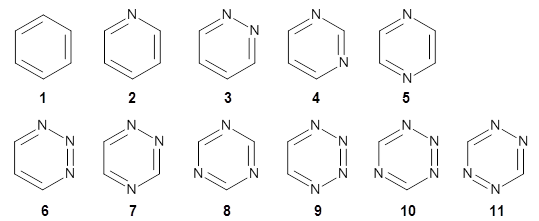
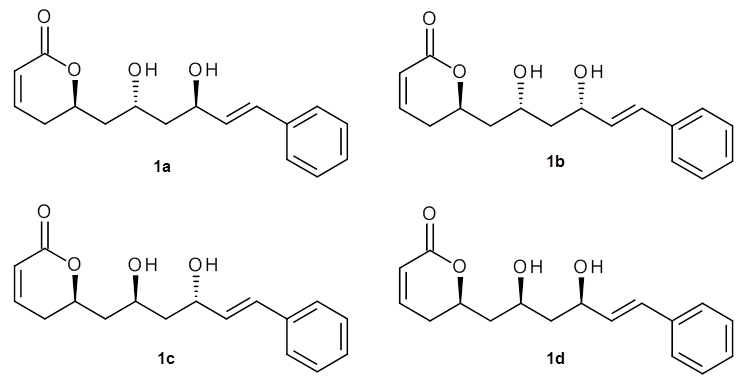
As a test case, they looked at cryptomoscatone D1 and D2 (1), for which the structures were determined with traditional methods. DP4 predicts that both cryptomoscatone D1 and D2 are structure 1d. However, DP4+ correctly predicts that cryptomoscatone D1 is 1b and cryptomoscatone D2 is 1a.
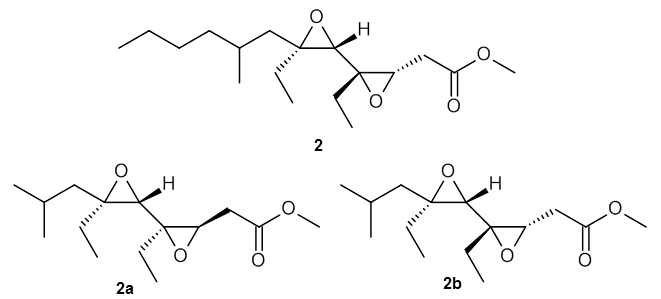
Lin and Tagliatatela-Scafati have reported the use of DP4+ to aid in the structure determination of plakdiepoxide 2.3 ROESY NMR could not provide definitive judgement of the stereochemical relationship about the bond between the two epoxide rings. They computed a number of conformers of the model compounds 2a and 2b at B3LYP/6-31G(d). The computed chemical shifts were then used with the DP4+ procedure to determine that the structure has the stereochemistry of 2b.
References
(1) Smith, S. G.; Goodman, J. M. "Assigning Stereochemistry to Single Diastereoisomers by GIAO NMR Calculation: The DP4 Probability," J. Am. Chem. Soc. 2010, 132, 12946-12959, DOI: 10.1021/ja105035r.
(2) Grimblat, N.; Zanardi, M. M.; Sarotti, A. M. "Beyond DP4: an Improved Probability
for the Stereochemical Assignment of Isomeric Compounds using Quantum Chemical Calculations of NMR Shifts," J. Org. Chem. 2015, 80, 12526-12534, DOI: 10.1021/acs.joc.5b02396.
(3) Chianese, G.; Yu, H.-B.; Yang, F.; Sirignano, C.; Luciano, P.; Han, B.-N.; Khan, S.; Lin, H.-W.; Taglialatela-Scafati, O. "PPAR Modulating Polyketides from a Chinese Plakortis simplex and Clues on the Origin of Their Chemodiversity," J. Org. Chem. 2016, 81 (12), 5135–5143, DOI: 10.1021/acs.joc.6b00695.
InChIs
Cryptomoscatone D1: InChI=1S/C17H20O4/c18-14(10-9-13-5-2-1-3-6-13)11-15(19)12-16-7-4-8-17(20)21-16/h1-6,8-10,14-16,18-19H,7,11-12H2/b10-9+/t14-,15-,16-/m1/s1
InChIKey=GOQOIZFMLWZVMB-OUUZNBFFSA-N
Cryptomoscatone D2: InChI=1S/C17H20O4/c18-14(10-9-13-5-2-1-3-6-13)11-15(19)12-16-7-4-8-17(20)21-16/h1-6,8-10,14-16,18-19H,7,11-12H2/b10-9+/t14-,15+,16+/m0/s1
InChIKey=GOQOIZFMLWZVMB-GLBZDCTLSA-N
plakdiepoxide: InChI=1S/C18H32O4/c1-6-9-10-13(4)12-17(7-2)16(22-17)18(8-3)14(21-18)11-15(19)20-5/h13-14,16H,6-12H2,1-5H3/t13?,14-,16-,17+,18-/m0/s1
InChIKey=YZQCELNTSCPAPG-KSZMJELXSA-N