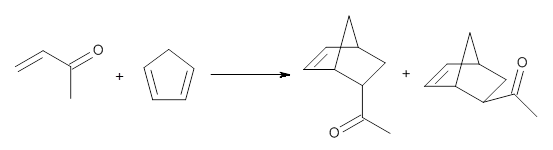
I discuss the aqueous Diels-Alder reaction in Chapter 7.1 of my book. A key case is the reaction of methyl vinyl ketone with cyclopentadiene, Reaction 1. The reaction is accelerated by a factor of 740 in water over the rate in isooctane.1 Jorgensen argues that this acceleration is due to stronger hydrogen bonding to the ketone than in the transition state than in the reactants.2-4
|
|
Rxn 1 |
Doubleday and Houk5 report a procedure for calculating trajectories including explicit water as the solvent and apply it to Reaction 1. Their process is as follows:
- Compute the endo TS at M06-2X/6-31G(d) with a continuum solvent.
- Equilibrate water for 200ps, defined by the TIP3P model, in a periodic box, with the transition state frozen.
- Continue the equilibration as in Step 2, and save the coordinates of the water molecules after every addition 5 ps, for a total of typically 25 steps.
- For each of these solvent configurations, perform an ONIOM computation, keeping the waters fixed and finding a new optimum TS. Call these solvent-perturbed transition states (SPTS).
- Generate about 10 initial conditions using quasiclassical TS mode sampling for each SPTS.
- Now for each the initial conditions for each of these SPTSs, run the trajectories in the forward and backward directions, typically about 10 of them, using ONIOM to compute energies and gradients.
- A few SPTS are also selected and water molecules that are either directly hydrogen bonded to the ketone, or one neighbor away are also included in the QM portion of the ONIOM, and trajectories computed for these select sets.
The trajectory computations confirm the role of hydrogen bonding in stabilizing the TS preferentially over the reactants. Additionally, the trajectories show an increasing asynchronous reactions as the number of explicit water molecules are included in the QM part of the calculation. Despite an increasing time gap between the formation of the first and second C-C bonds, the overwhelming majority of the trajectories indicate a concerted reaction.
References
(1) Breslow, R.; Guo, T. "Diels-Alder reactions in nonaqueous polar solvents. Kinetic
effects of chaotropic and antichaotropic agents and of β-cyclodextrin," J. Am. Chem. Soc. 1988, 110, 5613-5617, DOI: 10.1021/ja00225a003.
(2) Blake, J. F.; Lim, D.; Jorgensen, W. L. "Enhanced Hydrogen Bonding of Water to Diels-Alder Transition States. Ab Initio Evidence," J. Org. Chem. 1994, 59, 803-805, DOI: 10.1021/jo00083a021.
(3) Chandrasekhar, J.; Shariffskul, S.; Jorgensen, W. L. "QM/MM Simulations for Diels-Alder
Reactions in Water: Contribution of Enhanced Hydrogen Bonding at the Transition State to the Solvent Effect," J. Phys. Chem. B 2002, 106, 8078-8085, DOI: 10.1021/jp020326p.
(4) Acevedo, O.; Jorgensen, W. L. "Understanding Rate Accelerations for Diels−Alder Reactions in Solution Using Enhanced QM/MM Methodology," J. Chem. Theor. Comput. 2007, 3, 1412-1419, DOI: 10.1021/ct700078b.
(5) Yang, Z.; Doubleday, C.; Houk, K. N. "QM/MM Protocol for Direct Molecular Dynamics of Chemical Reactions in Solution: The Water-Accelerated Diels–Alder Reaction," J. Chem. Theor. Comput. 2015,

Henry Rzepa responded on 22 Feb 2016 at 1:39 am #
I want to raise the topic of RDM (research data management) and in particular how the research data produced by the simulation study described above might be shared/published.
Unlike many a quantum mechanical modelling of a reaction, where the calculation keywords (basis set, energy method) + final optimised coordinates are often enough to ensure reproducibility, the molecular dynamics experiment is instead really defined by a procedural workflow. We get some idea of the workflow from the seven point procedure outlined in the post above. I checked to see how this workflow appears in the supporting information (SI), and it largely consists of a number of Python scripts encapsulating the workflows, along with some distribution graphs. The “data” therefore consists of procedures rather than the data-rich outcomes of these procedures. This type of SI (unusually short by the standards of today at only 19 pages) is really quite different from other types of research “data” encountered in eg synthetic chemistry. In one regard it has the same characteristics, the SI is designed for re-use by a human. Such a human has to read and recognise the structure of the PDF document that constitutes the SI, edit it into scripts and other more semantically rich objects, and then submit these objects to the appropriately configured software components that are presumed installed. I suspect there are few specialists who would immediately have the technical savvy to perform this task quickly and without too many errors.
Of course, ideally we would like to be in a position where some software agent could instead be pointed at the SI, work out automatically what the semantics are and then re-run the simulations. It would also be a challenge to establish statistically whether the resulting outputs were or were not consistent with the conclusions of the original report. This utopian ideal is some way a way yet.
But I suggest it is important to think about these aspects. We are indeed moving into the era of non-statistical dynamics experiments for studying organic reactions, where perhaps in the next decade or so, most mechanistic explorations will be conducted in such a manner, and where reproducibility of results becomes an important consideration. How this is handled in the sense of publication of both the narrative and its associated research data is going to be the next great challenge in the scientific process.