Consider bicorannulenyl 1. Each corranulene unit is a bowl and each is chiral due to being monosubstituted. Additional chirality is due to the arrangement of the bowls along the C1-C1’ bond, the bond where the two rings join. So both rotation about the C1-C1’ bond and bowl inversion will change the local chirality (Scheme 1 distinguishes these two processes.) One might anticipate that the stereodynamics of 1 will be complicated!
Scheme 1

Rabinovitz, Scott, Shenhar and their groups have tackled the stereodynamics of 1.1 (This is a very nice joint experimental and theoretical study and I wonder why it did not appear in JACS.) At room temperature and above, one observes a single set of signals, a singlet and eight doublets, in the 1H NMR. Below 200 K, there are three sets of signals, evidently from three different diastereomers.
Each ring can have either P or M symmetry. The authors designate conformations using these symbols for each ring along with the value of the torsion angle about the C1-C1’ bond. So, for example, a PP isomer is the enantiomer of the MM conformer when their dihedral angles are of opposite sign.
DFT computations help make sense of these results. PBE0/6-31G* computations reveal all local minima and rotational and inversion transition states of 1. The lowest (free) energy structure is PP44 (see Figure 1). A very small barrier separates it from PP111; this barrier is related to loss of conjugation between the two rings. Further rotation must cross a much larger barrier (nearly 17 and 20 kcal mol-1). These barriers result from the interaction of the C2 hydrogen of one ring with either the C2’ or C10’ hydrogen, similar to the rotational barrier in 1,1’-binaphthyl. However, the barrier is lower in 1 than in 1,1’-binaphthyl due to the non-planar nature of 1 that allows the protons to be farther apart in the TS. Once over these large barriers, two local minima, PP-45 and PP-137, separated by a small barrier are again found.
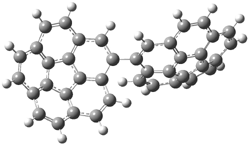 PP44 |
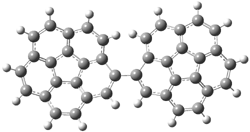 PP-28 |
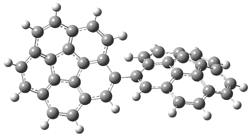 PP-45 |
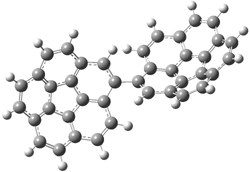
PP111 |
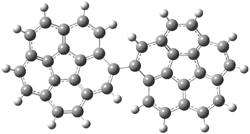
PP166 |
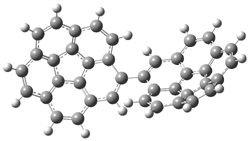
PP-137 |
Figure 1. PBE0/6-31G* optimized geometries and relative free energies (kcal mol-1) of the PP local minima (PP44, PP111, PP-45, and PP-137) and rotational transition states.1
The lowest energy bowl inversion transition state (P49) lies 9.6 kcal mole-1 above PP44. It is shown in Figure 2. The bowl inversion barrier is comparable to that found in other substituted corannulenes,2 which are typically about 9-12 kcal mol-1).
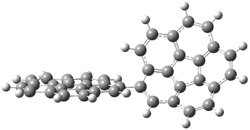
P49 |
Figure 2. PBE0/6-31G* optimized geometry and relative free energy of the lowest energy bowl inversion transition state.1
Interestingly, it is easier to invert the bowl than to rotate about the C1-C1’ bond. And this offers an explanation for the experimental 1H NMR behavior. At low temperature, crossing the low rotational barriers associated with loss of conjugation occurs. So, using the examples from Figure 1, PP44 and PP111 appear as a single time-averaged signal in the NMR. This leads to three pairs of enantiomers (S.PP/R.PP, S.MM/R.PP, and S.PM/R.MP), giving rise to the three sets of NMR signals.
References
(1) Eisenberg, D.; Filatov, A. S.; Jackson, E. A.; Rabinovitz, M.; Petrukhina, M. A.; Scott, L. T.; Shenhar, R., "Bicorannulenyl: Stereochemistry of a C40H18 Biaryl Composed of Two Chiral Bowls," J. Am. Chem. Soc. 2008, 73, 6073-6078, DOI: 10.1021/jo800359z.
(2) Wu, Y. T.; Siegel, J. S., "Aromatic Molecular-Bowl Hydrocarbons: Synthetic Derivatives, Their Structures, and Physical Properties," Chem. Rev. 2006, 106, 4843-4867, DOI: 10.1021/cr050554q.
InChIs
1: InChIKey = XZISQKATUXKXQW-UHFFFAOYAG
