In this post I cover two papers discussing application of computed NMR chemical shifts to structure identification and (yet) another review of computational techniques towards NMR structure prediction.
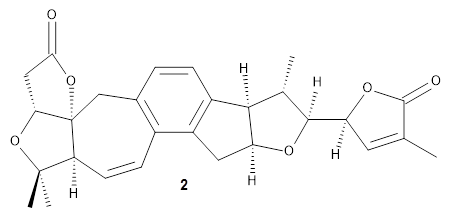
Grimblat, Kaufman, and Sarotti1 take up the structure of rubriflordilactone B 1, which was isolated from Schisandra rubriflora. The compound was then synthesized and its x-ray structure reported, however its NMR did not match with the natural extract. It was suggested that there were actually two compounds in the extract, the minor one was less soluble and is the crystallized 1, and a second compound responsible for the NMR signal.
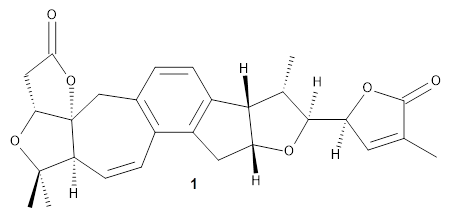
The authors looked at all stereoisomers of this molecule keeping the three left-most rings intact. The low energy rotamers of these 32 stereoisomers were then optimized at B3LYP/6-31G* and the chemical shifts computed at PCM(pyridine)/mPW1PW91/6-31+G**. To benchmark the method, DP4+ was used to identify which stereoisomer best matches with the observed NMR of authentic 1; the top fit (92.6% probability) was the correct structure.
The 32 stereoisomers were then tested against the experimental NMR of the natural extract. DP4+ with just the proton shifts suggested structure 2 (99.8% probability); however, the 13C chemical shifts predicted a different structure. Re-examination of the reported chemical shifts identifies some mis-assigned signals, which led to a higher C-DP4+ prediction. When all 128 stereoisomers were tested, structure 2 had the highest DP4+ prediction (99.5%), but the C-DP4+ prediction remained problematic (10.8%). Analyzing the geometries of all reasonable alternative for agreement with the NOESY spectrum confirmed 2. These results underscore the importance of using all data sources.
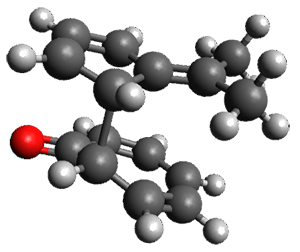
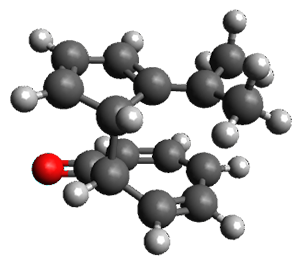
Reddy and Kutateladze point out the importance of using coupling constants along with chemical shifts in structure identification.2 They examined cordycepol A 3, obtained from Cordyceps ophioglossoides. They noted that the computed chemical shifts and coupling constants of originally proposed structure 3a differed dramatically from the experimental values.
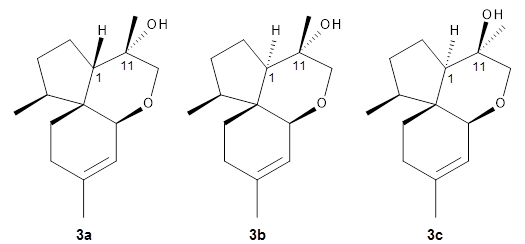
They first proposed that the compound has structure 3b. The computed coupling constants using their relativistic force field.3 The experimental coupling constants for the proton H1 are 13.4 and 7.1 Hz. The computed values for 3a are 8.9 and 1.6 Hz, and this structure is clearly incorrect. The coupling constants are improved with 3b, but the 13C chemical shifts are in poor agreement with experiment. So, they proposed structure 3c, the epimer at both C1 and C11 of the original structure.
They optimized four conformations of 3c at B3LYP/6-31G(d) and obtained Boltzmann-weighted chemical shifts at mPW1PW91/6-311+G(d,p). The RMS deviation of the computed 13C chemical shifts relative to the experiment is only 1.54 ppm, and more importantly, the computed coupling constants of 13.54 and 6.90 Hz are in excellent agreement with the experiment values.
Lastly, Grimblat and Sarotti present a review of a number of methods for using computed NMR chemical shifts towards structure prediction.4 These methods include CP3, DP4, DP4+ (all of which I have posted on in the past) and an artificial neural network approach of their own design. They discuss a number of interesting cases where each of these methods has been crucial in identifying the correct chemical structure.
References
1. Grimblat, N.; Kaufman, T. S.; Sarotti, A. M., "Computational Chemistry Driven Solution to Rubriflordilactone B." Org. Letters 2016, 18, 6420-6423, DOI: 10.1021/acs.orglett.6b03318.
2. Reddy, D. S.; Kutateladze, A. G., "Structure Revision of an Acorane Sesquiterpene Cordycepol A." Org. Letters 2016, 18, 4860-4863, DOI: 10.1021/acs.orglett.6b02341.
3. (a) Kutateladze, A. G.; Mukhina, O. A., "Minimalist Relativistic Force Field: Prediction of Proton–Proton Coupling Constants in 1H NMR Spectra Is Perfected with NBO Hybridization Parameters." J. Org. Chem. 2015, 80, 5218-5225, DOI: 10.1021/acs.joc.5b00619; (b) Kutateladze, A. G.; Mukhina, O. A., "Relativistic Force Field: Parametrization of 13C–1H Nuclear Spin–Spin Coupling Constants." J. Org. Chem. 2015, 80, 10838-10848, DOI: 10.1021/acs.joc.5b02001.
4. Grimblat, N.; Sarotti, A. M., "Computational Chemistry to the Rescue: Modern Toolboxes for the Assignment of Complex Molecules by GIAO NMR Calculations." Chem. Eur. J. 2016, 22, 12246-12261, DOI: h10.1002/chem.201601150.
InChIs
1: InChI=1S/C28H30O6/c1-13-9-20(32-26(13)30)25-14(2)24-17-6-5-15-12-28-21(8-7-16(15)18(17)10-19(24)31-25)27(3,4)33-22(28)11-23(29)34-28/h5-9,14,19-22,24-25H,10-12H2,1-4H3/t14-,19+,20-,21-,22+,24-,25-,28+/m0/s1
InChIKey=JGSLSHOXBXVVTQ-NEUKEVNNSA-N
2: InChI=1S/C28H30O6/c1-13-9-20(32-26(13)30)25-14(2)24-17-6-5-15-12-28-21(8-7-16(15)18(17)10-19(24)31-25)27(3,4)33-22(28)11-23(29)34-28/h5-9,14,19-22,24-25H,10-12H2,1-4H3/t14-,19-,20-,21-,22+,24+,25-,28+/m0/s1
InChIKey=JGSLSHOXBXVVTQ-WQIRXNRDSA-N
3c: InChI=1S/C16H28O2/c1-6-11(2)9-14-16(5)12(3)7-8-13(16)15(4,17)10-18-14/h9,12-14,17H,6-8,10H2,1-5H3/b11-9-/t12-,13-,14-,15-,16+/m0/s1
InChIKey=WPQIVUHVYBQTBG-AWEVENECSA-N