Antiaromatic compounds by definition are unstable and so difficult to prepare. One approach to increase their stability is to fuse aromatic ring(s) onto the antiaromatic system. I discuss in this blog post two different scaffolds where this approach has been successful in preparing molecules that express some degree of antiaromaticity. In addition, I mention a technique to aid in evaluating the aromatic/antiaromatic character.
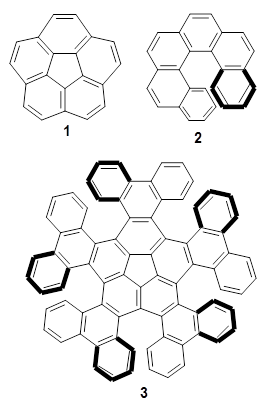
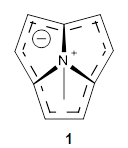
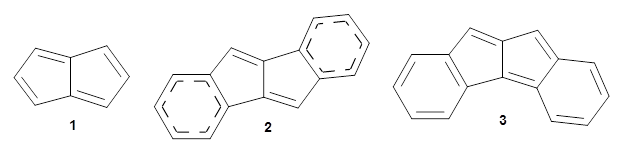
Pentalene 1 is a formal 8-π electron system and would be antiaromatic. To avoid this antiaromatic character, the double bonds are localized. Fusing benzenoid rings to pentalene to give dibenzo[a,e]penatalene 2 has been done, but the central rings avoid antiaromatic character by expressing the Kekule structure shown below.
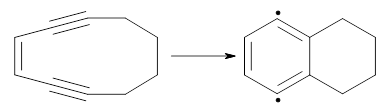
Yasuda and coworkers report the preparation of mesityl-substituted dibenzo[a,f]penatalene
3.1 Resonance structures of 3¸ shown below, either have only one aromatic ring, or have two aromatic rings along with a trimethylenemethane (TMM) diradical component. Thus, one might expect 3 to express more antiaromatic character than 2.
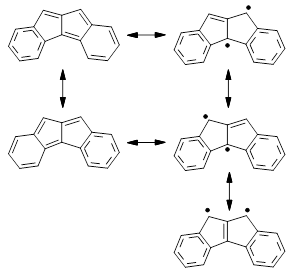
NICS(1) values, computed at B3LYP/6-31G**, for 2 are -6.23 for the 6-member ring and +5.87 ppm for the 5-member ring, showing reduced aromaticity of the former ring. In sharp contrast, the NICS(1) values for 3 are +7.48 for the 6-member ring and +25.5 ppm for the 5-member ring, indicating substantial antiaromatic character for both rings. The calculated spin density distribution shows largest unpaired density on the expected carbon atoms based on the resonance structures involving the TMM fragment.
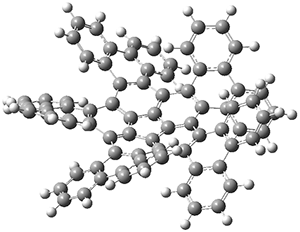
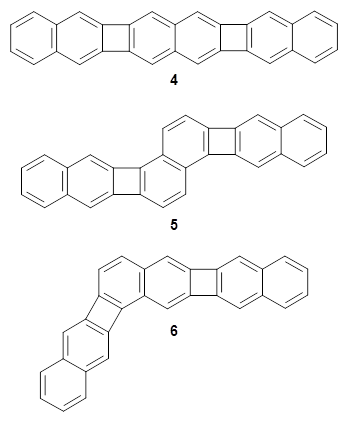
Xia and coworkers have prepared substituted analogues of the three structural isomers whereby three naphthylene units are fused together creating two cyclobutadienoid rings.2 These three frameworks are molecules 4-6. The 4-member rings are formally antiaromatic, tempered by the fused aromatic naphthylene groups. The question is then how does the different attachment geometry manifest in aromatic and/or antiaromatic character?
The computations take advantage of the NICS-XY method – well, a variation of this method. I had meant to write a post about the NICS-XY method when Stanger published it,3 but I just never got around to it. The idea is that NICS is evaluated typically at a single point, and just which point to use has been the subject of some discussion. Instead, Stanger proposes the NICS-XY method as a grid of points perpendicular to the plane of the molecule, typically in the plane bisecting the molecule. Trends in the values as one moves across the ring and perpendicular to the ring could assist in identifying aromatic/antiaromatic behavior.
Xia computed the NICSπZZ along a line in the molecular plane bisecting the rings. This is shown in the figure below, which I have reproduced from the article. For example, for 4, which is compound 1 in the Xia paper and the figure below, the NICS values are taken along the line that horizontally bisects the molecule. In ring A, the values are negative, indicative of an aromatic ring. Across ring B, the values are still negative, but not as negative as for ring A, indicating a diminished aromaticity. In ring C, the values are positive, as one would expect for the antiaromatic cyclobutadienoid ring.
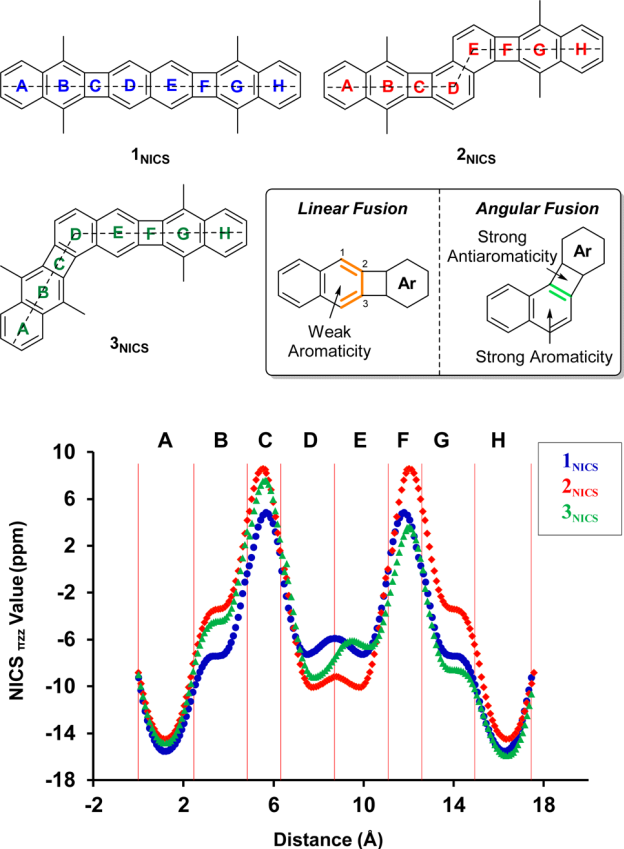
Figure taken from J. Am. Chem. Soc. 2017, 139, 15933-15939.
The authors highlight two trends. First, in the linear fusion (see the inset above), the aromatic ring fused to the cyclobutadienoid ring expresses diminished aromaticity. This can be understood in the following way. In naphthalene, the C2-C3 bond is longer than the C1-C2 bond. When the cyclobutadienoid is fused at the C2-C3 bond, it can lengthen even more to weaken the antiaaromaticity of the 4-member ring, and this consequently reduces the aromaticity of the 6-member ring. Fusion of the cyclobutadienoid ring at C1-C2, the shorter bond, causes a higher degree of antiaromaticity in the 4-member ring. The lengthening of this C1-C2 bond to try to reduce the antiromaticity of the 4-member ring leads to greater bond equalization in the 6-member ring, and its consequently greater aromatic character.
References
1. Konishi, A.; Okada, Y.; Nakano, M.; Sugisaki, K.; Sato, K.; Takui, T.; Yasuda, M., "Synthesis and Characterization of Dibenzo[a,f]pentalene: Harmonization of the Antiaromatic and Singlet Biradical Character." J. Am. Chem. Soc. 2017, 139, 15284-15287, DOI: 10.1021/jacs.7b05709.
2. Jin, Z.; Teo, Y. C.; Teat, S. J.; Xia, Y., "Regioselective Synthesis of [3]Naphthylenes and Tuning of Their Antiaromaticity." J. Am. Chem. Soc. 2017, 139, 15933-15939, DOI: 10.1021/jacs.7b09222.
3. Gershoni-Poranne, R.; Stanger, A., "The NICS-XY-Scan: Identification of Local and Global Ring Currents in Multi-Ring Systems." Chem. Eur. J. 2014, 20, 5673-5688, DOI: 10.1002/chem.201304307.
InChIs
1: InChI=1S/C8H6/c1-3-7-5-2-6-8(7)4-1/h1-6H
InChIKey=GUVXZFRDPCKWEM-UHFFFAOYSA-N
2: InChI=1S/C16H10/c1-3-7-13-11(5-1)9-15-14-8-4-2-6-12(14)10-16(13)15/h1-10H
InChIKey=OZEPXROCWSMGGM-UHFFFAOYSA-N
3: InChI=1S/C16H10/c1-3-7-14-11(5-1)9-13-10-12-6-2-4-8-15(12)16(13)14/h1-10H
InChIKey=XOERMEAUYMRNNZ-UHFFFAOYSA-N
4: InChI=1S/C30H16/c1-2-6-18-10-24-23(9-17(18)5-1)27-13-21-15-29-25-11-19-7-3-4-8-20(19)12-26(25)30(29)16-22(21)14-28(24)27/h1-16H
InChIKey=CHDMCKMZQIHGAH-UHFFFAOYSA-N
5: InChI=1S/C30H16/c1-3-7-19-15-27-25(13-17(19)5-1)23-11-9-22-21(29(23)27)10-12-24-26-14-18-6-2-4-8-20(18)16-28(26)30(22)24/h1-16H
InChIKey=LPXGODOTGXTPRU-UHFFFAOYSA-N
6: InChI=1S/C30H16/c1-2-7-19-13-26-25(12-18(19)6-1)27-15-21-9-10-22-24-11-17-5-3-4-8-20(17)14-29(24)30(22)23(21)16-28(26)27/h1-16H
InChIKey=BKMGPFRQJXDFJQ-UHFFFAOYSA-N