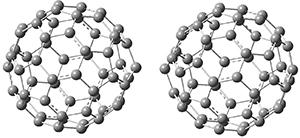
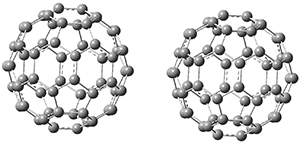
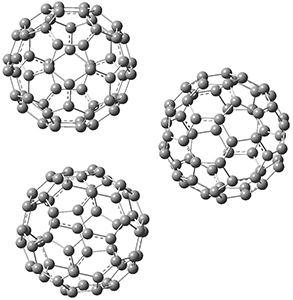
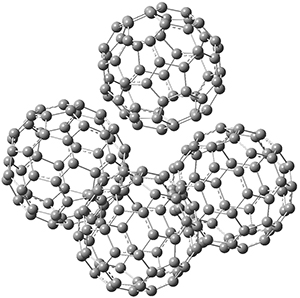
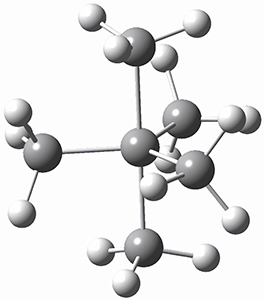
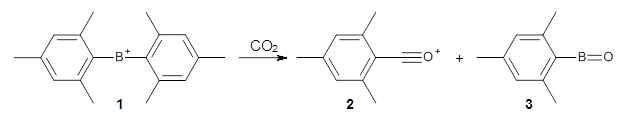
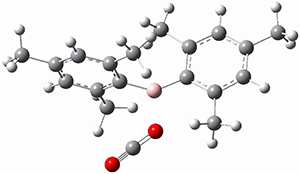
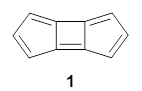
1 is notable for its very short central C-C bond, computed at B1B95/6-31G(d) to be only 1.30 Å. Also notable is that 1 can rearrange to the carbene 2 with a release of considerable energy (ΔE=-105.4 kcal mol-1). Nonetheless, the barrier for this rearrangement is 6.7 kcal mol-1 suggesting that 1 might be stable and isolable at low temperatures. (See this previous post for more discussion on this rearrangement, including interactive molecules.)
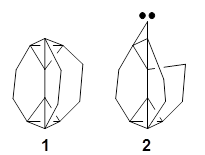
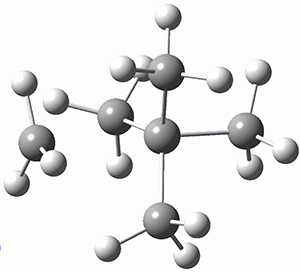
Kozuch has now examined this rearrangement in more detail, to see if 1 is really stable.1 The issue he raises is the role of quantum mechanical tunneling – since the distance that the carbon atoms need to move in reaching the TS is rather small, perhaps heavy atom tunneling might manifest. In the absence of tunneling, conventional variation transition state theory (CVT) predicts that the half-life of 1 is 170 s at 75 K, and longer still at even lower temperatures. However, the situation is radically different when tunneling is included. Accounting for tunneling using the small curvature tunneling (SCT) approximation predicts a half-life of 1.6 x 10-3 s at 75 K and only a minimally longer half-life of 4.6 x 10-3s at 10 K. Thus, Kozuch concludes that 1 is not stable at any temperature! One should thus be cautious in applying the term “stable” to a compound that might be quite strained and susceptible to tunneling.
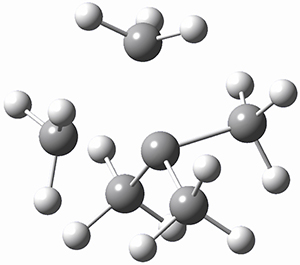
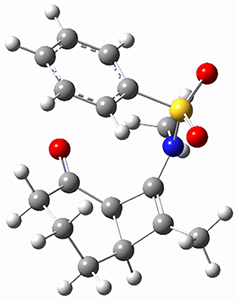
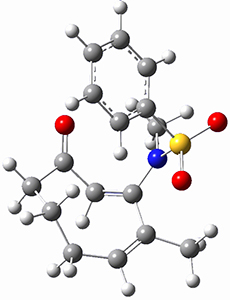
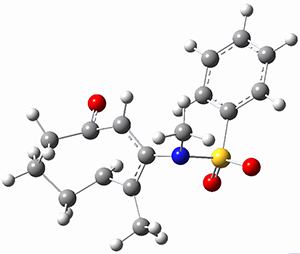
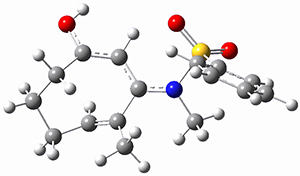
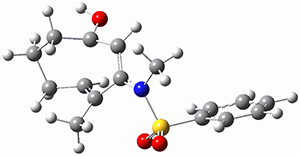
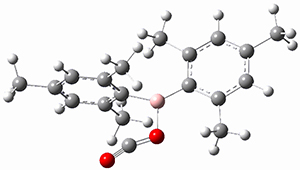
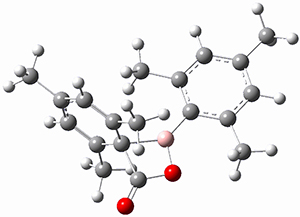
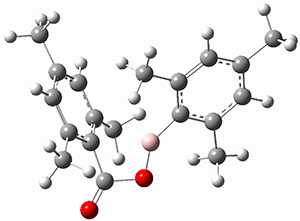
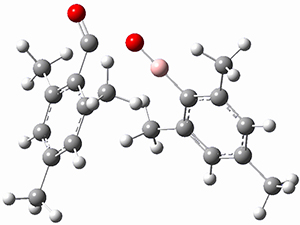
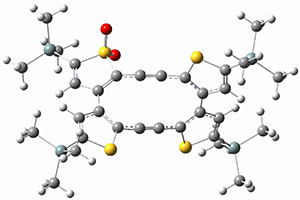
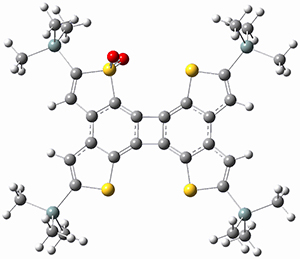
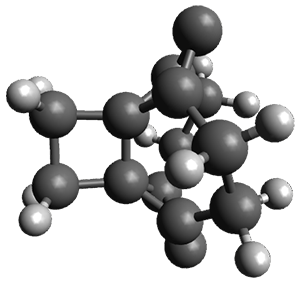
(As an aside, Kozuch also notes that 2 can rearrange into 3 and this rearrangement also has a very short half-life on the order of milliseconds at cryogenic temperatures. The structure of 3 is shown in Figure 1.)
|
Figure 1. B1B95/6-31G(d) optimized structure of 3.
References
1) Kozuch, S. “A Quantum Mechanical “Jack in the Box”: Rapid Rearrangement of a Tetrahedryl-Tetrahedrane via Heavy Atom Tunneling,” Org. Lett., 2014, 16, 4102-4105, DOI: 10.1021/ol5017977.
InChIs
1: InChI=1S/C14H12/c1-2-8-11-5-3-9-7(1)10(9)4-6-12(8,11)14(8,11)13(7,9)10/h1-6H2
InChIKey=LNBZAENQMFDBJW-UHFFFAOYSA-N
2: InChI=1S/C14H12/c1-3-11-12-4-2-9-7-8(1,9)10(9)5-6-13(11,12)14(10,11)12/h1-6H2
InChIKey=UKVODHRLGFPZPT-UHFFFAOYSA-N
3: InChI=1S/C14H12/c1-2-10-8-12(10)4-3-11-7-9(1,11)13(11)5-6-14(10,12)13/h1-6H2
InChIKey=AATRYSJRWFKWAY-UHFFFAOYSA-N