An alternative take on the nature of the interaction in π-stacking is offered by Wheeler and Houk.1 They start by examining the binding between benzene and a series of 24 substituted benzenes. Two representative dimmers are shown in Figure 1, where the substituent is NO2 or CH2OH. As was noted in a number of previous studies,2-6 the binding with any substituted benzene is stronger than the parent benzene dimer. Nonetheless, Wheeler and Houk point out that the binding energy has a reasonable correlation with σm. It appears that the benzene dimer itself is the outlier; the binding energy when the substituent is CH2OH, whose σm value is zero, is bound more tightly than the benzene dimer. They conclude that there is a dispersive interaction between any substituent and the other benzene ring.
|
(a) 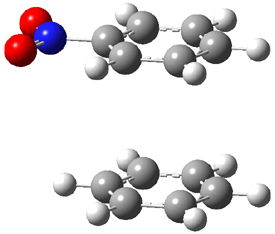 |
(b) 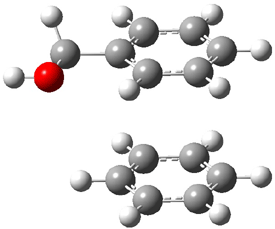 |
|
(c) 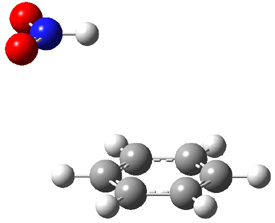 |
(d) 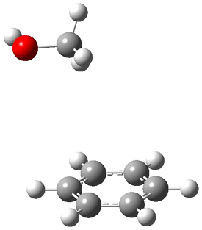 |
Figure 1. MO5-2X/6-31+G(d) optimized geometries of (a) C6H6-C6H5NO2, (b) C6H6-C6H5CH2OH, (c) C6H6-HNO2, and (d) C6H6-HCH2OH.1
They next constructed an admittedly very crude model system whereby the substituted benzene C6H5X is replaced by HX; the corresponding models are also shown in Figure 1. The binding energies of these model dimmers correlates very well with the real dimmers, with r = 0.91. Rather than involving the interaction of the π-electrons, the origin of the enhanced binding in aromatic dimers involves electrostatic interactions of the substituent with the other aromatic ring – effectively the quadrupole of the unsubstituted ring interacts with the dipoles of the substituent and its ring system. In addition, the inherent dispersive interaction increase the binding.
References
(1) Wheeler, S. E.; Houk, K. N., "Substituent Effects in the Benzene Dimer are Due to Direct Interactions of the Substituents with the Unsubstituted Benzene," J. Am. Chem. Soc., 2008, 130, 10854-10855, DOI: 10.1021/ja802849j.
(2) Sinnokrot, M. O.; Sherrill, C. D., "Unexpected Substituent Effects in Face-to-Face π-Stacking Interactions," J. Phys. Chem. A, 2003, 107, 8377-8379, DOI: 10.1021/jp030880e.
(3) Sinnokrot, M. O.; Sherrill, C. D., "Substituent Effects in π-&pi Interactions: Sandwich and T-Shaped Configurations," J. Am. Chem. Soc., 2004, 126, 7690-7697, DOI: 10.1021/ja049434a.
(4) Sinnokrot, M. O.; Sherrill, C. D., "Highly Accurate Coupled Cluster Potential Energy Curves for the Benzene Dimer: Sandwich, T-Shaped, and Parallel-Displaced Configurations," J. Phys. Chem. A, 2004, 108, 10200-10207, DOI: 10.1021/jp0469517
(5) Lee, E. C.; Kim, D.; Jurecka, P.; Tarakeshwar, P.; Hobza, P.; Kim, K. S., "Understanding of Assembly Phenomena by Aromatic-Aromatic Interactions: Benzene Dimer and the Substituted Systems," J. Phys. Chem. A 2007, 111, 3446-3457, DOI: 10.1021/jp068635t.
(6) Grimme, S.; Antony, J.; Schwabe, T.; Mück-Lichtenfeld, C., "Density functional theory with dispersion corrections for supramolecular structures, aggregates, and complexes of
(bio)organic molecules," Org. Biomol. Chem. 2007, 741-758, DOI: 10.1039/b615319b

Computational Organic Chemistry » Benzene dimer responded on 27 Jan 2009 at 10:17 am #
[…] Hobza1 has published a very high-level computational study of the benzene dimer as a benchmark for this model of π-π stacking – a topic I have touched upon a number of times in this blog (post 1, post 2) . There are four local energy minima, shown in Figure 1. The most stable dimer is the tilted T-structure (TT), a structure often overlooked. Its complexation energy, computed at CCSD(T)/CBS, is 2.78 kcal mol-1. Only slightly higher in energy is the parallel displaced structure (PD), with a stabilization energy of 2.70 kcal mol-1. The T structure (T) is essentially isoenergetic with the PD one. The perfectly stacked structure (S) is much less stable, with a dimerization energy of 1.64 kcal mol-1. The DTF-D method, using the BLYP functional with dispersion parameters optimized for the benzene dimer provide energies within 0.2 kcal mol-1 of the computationally much more expensive benchmark values. As a word of caution though: use of more general dispersion parameters gives energies far worse and predicts the wrong energy order of the dimers. […]
Bright Emenike responded on 07 Dec 2010 at 2:02 pm #
Gung & Co workers examined Houk’s claim experimentally by using a set of triptycene based conformationally reporters. In their study, they revealed that the observed trend due to substittuent effect in the Wheeler & Houks’s model HX-benzene complex may be be due H…Benzene interaction instead of X…Benzene (direct Interaction) as depicted on their publication. Read more: “Quantification of CH⋅⋅⋅π Interactions: Implications on How Substituent Effects Influence Aromatic Interactions” Chemistry – A European Journal
Volume 16, Issue 41, pages 12357–12362, November 2, 2010
Steven Bachrach responded on 07 Dec 2010 at 2:38 pm #
Thanks for this paper to my attention!
Here is a direct link to the Gung article: 10.1002/chem.201001362.