Unusual potential energy surfaces are a theme of this blog and my book (see chapter 7). Examples might include bifurcations and valley inflection points and often lead to unusual dynamics. Tantillo has now reported a bifurcation on the PES for terpene synthesis, specifically the pathway for synthesis of abietadiene.1
Tantillo discusses two possible cation rearrangement pathways. The first is pretty ordinary, but in the second, the precursor cation 1 can rearrange through either of two transition states 2a or 2b (Scheme 1). The IRC computation from 2a connects back to 1, but in the forward direction it connects to another transition state 3. This TS (3) connects products 4 and 5. These structures are drawn in Figure 1.
Thus, the potential energy surface displays a bifurcation, and one might expect unusual dynamic effects to operate.
Scheme 1

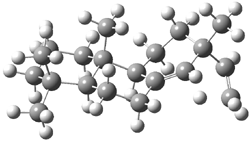 2a |
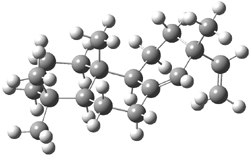 2b |
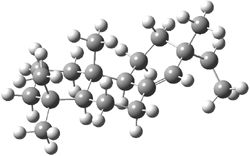 3 |
|
Figure 1. B3LYP/6-31+G(d,p) optimized transition structures of 2-3.1
References
(1) Hong, Y. J.; Tantillo, D. J., "A potential energy surface bifurcation in terpene biosynthesis," Nature Chem. 2009, 1, 384-389 DOI: 10.1038/nchem.287.

Henry Rzepa responded on 24 Sep 2009 at 3:39 am #
Bifurcated potential energy surfaces are an interesting, and relatively unexplored aspect of reaction mechanisms. We have found a few examples ourselves. The stationary points in such surfaces are particularly interesting, and I believe that only by inspecting the coordinates, and even better, the normal reaction modes, can one start to get genuine insight. So I was off to the supporting information associated with this article that I headed. It was a pleasant surprise to find most of the coordinates there, but less welcome to notice that the publisher had not exactly made it easy to use these coordinates. Thus almost all of the coordinates were page broken, with the publisher inserting headers into them to achieve the house style. Thus the potential user of this information receives a mixture of style and content, which they have to separate themselves, manually. They then have to add their own header.
A tad more distressing was that nowhere were the calculated normal modes accessible. Where comprehensive IRCs have been calculated, it is obviously important to have access to such information. The only option is to recalculate them, which can take hours (perhaps days) per compound. One would have to be highly motivated to do this (and have spare time on one’s HPC systems).
So why is it that a brand new chemistry journal, launched to offer a new improved service to chemists, has adopted the old tired format of offering supporting information as a contiguous Acrobat file? Perhaps because all the other publishers do this? Or that the readership expects it in this format? Its not as if perfectly viable alternatives have not been around for some time.
Let me ask another question. How many computational chemists have access to a local digital repository? Indeed one that can accept computational outputs for deposition? Might I venture to suggest that world-wide, the number is very tiny! Are they clamouring for their institution to install one on their behalf? Let me put Steve Bachrach on the spot here. He has often stated that for the purpose of creating his blog items, he almost always has to recalculate the relevant systems. He probably grinds his teeth at having to cope with Acrobat! But the task is accomplished and the blog article is published. But where is YOUR digital repository Steve? Have you asked your institution if one could be installed for the the purpose (inter alia) of supporting the blog? How can people retrieve data from your own blog?
I did try to extract some of the data (i.e. coordinates) from your blog Steve. Normally, a right mouse click on the Jmol may offer the opportunity to see the properties of the molecule (although actually downloading the coordinates to a local hard drive should only be possible if a signed Jmol applet has been deployed). But I was unable to in this case (I am not actually sure why).
Steve and I do care about data. But there as so many hurdles in the way of obtaining it, and the publishers currently put too little effort into solving these problems. Perhaps they cannot see the business case for doing so?
Anyway Steve, is there any chance of seeing some animated transition modes for 2a/2b/3 in your post?
Steven Bachrach responded on 25 Sep 2009 at 2:47 pm #
I have now upgraded the Jmol so that the right-click options that Henry mentions are available.
But I do not have the vibrational modes of the TSs mentioned in the article – I just grabbed the coordinates from the supporting information and I did not rerun the computations to generate these frequencies.
Stuart responded on 28 Sep 2009 at 9:37 am #
This may, perhaps, be the somewhat flippant answer, but an e-mail address is included for the corresponding author of this (and every other) contribution published in Nature Chemistry. Why not go to the horse’s mouth for said data – or any other data for that matter.
On a less flippant note, we’d be happy to host data sets, but we do not make it a requirement of submission (or publication). If authors send us data, we will host it. It’s not just publishers who need to get involved in making data accessible (which we do, I admit), it’s authors too.
Steven Bachrach responded on 28 Sep 2009 at 10:30 am #
As a flippant reply – what happens when the authors doen’t reply or moves to a new institution or (more permanently) dies? I fear that data sets are permanently lost!
But more seriously, I do completely agree that the blame for absent data sets or incomplete data sets is due to authors and readers! It si US who must scream at publishers that articles must contain the data upon which the artcice lies. Data sets must be deposited in useful formats (not the appropriately maligned Acrobat format that Henry rails about in his comment above). And these data sets must be made available to all for free, and with no restrictions upon the use and re-use. I am glas to see that Nature Chemistry is willing to partner with authors.
So I call upon all Nature Chemistry authors to take up Stuart’s gauntlet and deposited full data sets in usable formats.
Henry Rzepa responded on 30 Sep 2009 at 11:21 am #
To add to this (lively) discussion, I recently had to give a public lecture to an audience not composed entirely of chemists. I decided to show in this lecture some carbon tori, which had been published by a group of physicists (DOI: 10.1103/PhysRevLett.88.217206). Since these systems contained some 1400 carbon atoms, I was reluctant to try to build the molecule myself, and I hence did endeavour to contact the original authors. One of the students responded, but replied that the coordinates had been on an old computer, and were now lost. We are talking about an article published in 2002! I eventually identified another researcher in the field, who was able to generate the coordinates using a program he had written (and which I then re-optimized using molecular mechanics to remove some of the artefacts introduced by that program!). It was a stroke of luck, and not a little persistence on my part. The result, by the way, can be seen here. Oh, I should mention that I received the coordinates in a format I had never seen before, and had to put further effort into changing the format into something that (Jmol) could display.
So I do agree with what Steve writes. But a valid question to ask is “what is likely to decay first, the ability to contact authors of articles, or the supporting information published with an article”? In some cases, the latter of course is already mounted in a “decayed format” (ie one not immediately fit for the purpose of repeating the original work).