Here is another nice example of the partnership between experiment and computation in ascertaining molecular structure. The Sarpong, Tantillo, Andersen, Berlinck, and Miller groups collaborated on the synthesis, characterization and biosynthesis of some metabolites from Penniculium strains.1 I will focus here on just the structural identification component of this paper; the synthesis and the biosynthesis are very interesting too!
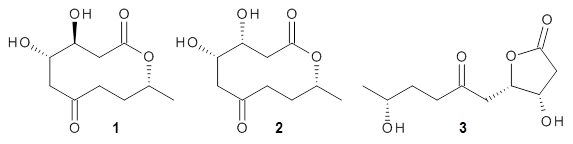
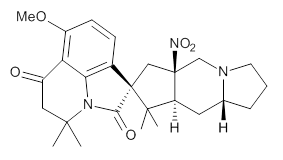
Cyclopiamine A 1 and cyclopiamine B 2 interconvert through an intermediate that allows for the epimerization at carbon bearing the nitro group.2
|
1
|
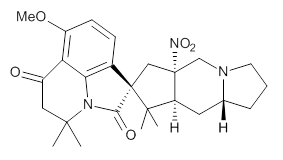
2
|
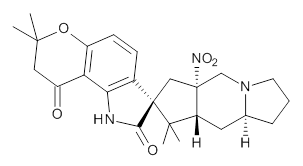
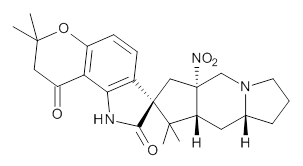
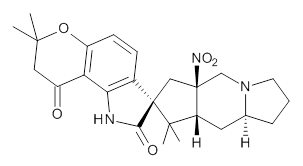
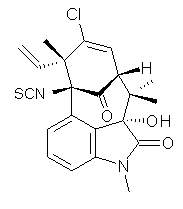
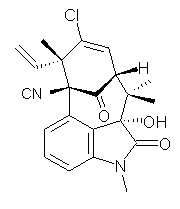
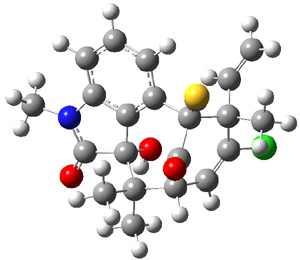
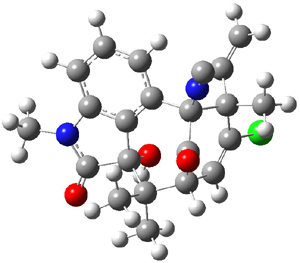
Citrinalin A 3 might also seem to undergo the same type of ring opening-ring closing reaction to produce citrinalin B. However, the original proposed structure3 of citrinalin B 4 implies an epimerization at a different carbon (at the ring fusion to the terminal 5 member ring). These authors suggested that perhaps the proper structure of citrinalin B is 5, which differs from citrinalin A only at the carbon bearing the nitro group, analogous to the relationship between 1 and 2.
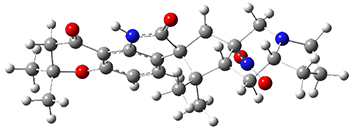
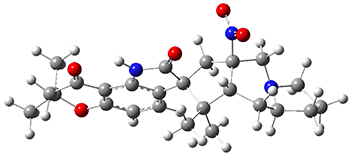
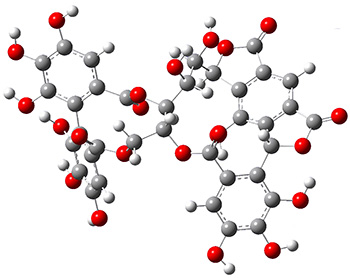
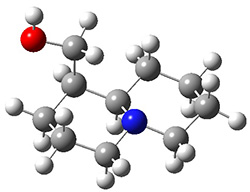
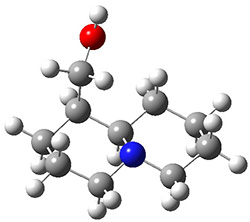
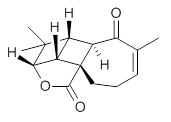
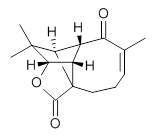
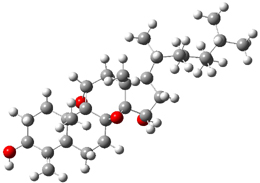
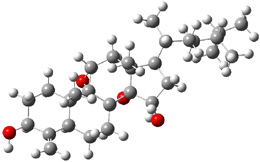
The low energy conformations of both 4 and 5 (actually the trifluoroacetic acid salts) were optimized at B3LYP/6-31+G(d,p) and the chemical shifts for both 1H and 13C were computed, Boltzmann-weighted and scaled, and then compared with the NMR spectra of authentic citrinalin B. (The lowest energy conformations of 4 and 5 are shown in Figure 1.) The corrected mean absolute deviations for the 1H and 13C chemical shift for the original structure 4 are 0.45 ppm and 2.0 ppm, respectively (with the largest outliers of 2.3 ppm for H and 9.6 ppm for C). These errors are about twice what is observed in comparing the experimental and computed 1H and 13C chemical shifts of 3. The agreement between the computed and experimental values using 5 are much improved, with mean deviations of 0.12 and 1.6ppm, and largest deviations of 0.38 ppm for 1H and 4.4 ppm for 13C. Use of Goodman’s DP4 method indicates a 100% probability that the structure of citrinalin B is 5. This prediction is confirmed by the x-ray structure.
Figure 1. B3LYP/6-31+G(d,p) optimized lowest energy conformers of 4 and 5.
References
(1) Mercado-Marin, E. V.; Garcia-Reynaga, P.; Romminger, S.; Pimenta, E. F.; Romney, D. K.; Lodewyk, M. W.; Williams, D. E.; Andersen, R. J.; Miller, S. J.; Tantillo, D. J.; Berlinck, R. G. S.; Sarpong, R. "Total synthesis and isolation of citrinalin and cyclopiamine congeners," Nature 2014, 509, 318-324, DOI: 10.1038/nature13273.
(2) Bond, R. F.; Boeyens, J. C. A.; Holzapfel, C. W.; Steyn, P. S. "Cyclopiamines A and B, novel oxindole metabolites of Penicillium cyclopium westling," J. Chem. Soc., Perkin Trans I 1979, 1751-1761, DOI: 10.1039/P19790001751.
(3) Pimenta, E. F.; Vita-Marques, A. M.; Tininis, A.; Seleghim, M. H. R.; Sette, L. D.; Veloso, K.; Ferreira, A. G.; Williams, D. E.; Patrick, B. O.; Dalisay, D. S.; Andersen, R. J.; Berlinck, R. G. S. "Use of Experimental Design for the Optimization of the Production of New Secondary Metabolites by Two Penicillium Species," J. Nat. Prod. 2010, 73, 1821-1832, DOI: 10.1021/np100470h.
InChIs
1: InChI=1S/C26H33N3O5/c1-23(2)12-17(30)20-18(34-5)9-8-16-21(20)28(23)22(31)26(16)13-25(29(32)33)14-27-10-6-7-15(27)11-19(25)24(26,3)4/h8-9,15,19H,6-7,10-14H2,1-5H3/t15-,19+,25+,26-/m1/s1
InChIKey=LUESCPPTDJZZQN-VBPSCBQFSA-N
2: InChI=1S/C26H33N3O5/c1-23(2)12-17(30)20-18(34-5)9-8-16-21(20)28(23)22(31)26(16)13-25(29(32)33)14-27-10-6-7-15(27)11-19(25)24(26,3)4/h8-9,15,19H,6-7,10-14H2,1-5H3/t15-,19+,25-,26-/m1/s1
InChIKey=LUESCPPTDJZZQN-DCWDXPITSA-N
3: InChI=1S/C25H31N3O5/c1-22(2)11-16(29)19-17(33-22)8-7-15-20(19)26-21(30)25(15)12-24(28(31)32)13-27-9-5-6-14(27)10-18(24)23(25,3)4/h7-8,14,18H,5-6,9-13H2,1-4H3,(H,26,30)/t14-,18+,24+,25-/m0/s1
InChIKey=QVBARITWSQCBBV-DTKOROOESA-N
4: InChI=1S/C25H31N3O5/c1-22(2)11-16(29)19-17(33-22)8-7-15-20(19)26-21(30)25(15)12-24(28(31)32)13-27-9-5-6-14(27)10-18(24)23(25,3)4/h7-8,14,18H,5-6,9-13H2,1-4H3,(H,26,30)/t14-,18-,24-,25+/m1/s1
InChIKey=QVBARITWSQCBBV-KUCRYEJDSA-N
5: InChI=1S/C25H31N3O5/c1-22(2)11-16(29)19-17(33-22)8-7-15-20(19)26-21(30)25(15)12-24(28(31)32)13-27-9-5-6-14(27)10-18(24)23(25,3)4/h7-8,14,18H,5-6,9-13H2,1-4H3,(H,26,30)/t14-,18+,24-,25-/m0/s1
InChIKey=QVBARITWSQCBBV-QBDGMKJCSA-N