A quick note here on the use of computed NMR to determine stereochemical structure. The Garg group synthesized two “oxidized welwitindolines”, compounds 1 and 2.1 The relative stereochemistry at the C3 position (the carbon with the hydroxy group) was unknown.
|
|
|
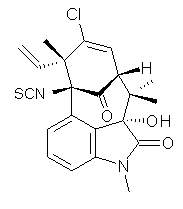
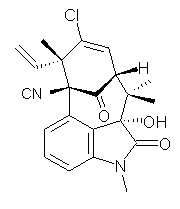
Low energy gas-phase conformers of both epimers of 1 and 2 were optimized at B3LYP/6-31+G(d,p). (These computations were done by the Tantillo group.) See Figure 1 for the optimized lowest energy conformers. Using these geometries the NMR chemical shifts were computed at mPW1PW91/6-311+G(d,p) with implicit solvent (chloroform). The chemical shifts were Boltzmann-weighted and scaled according to the prescription (see this post) of Jain, Bally and Rablen.2 The computed chemical shifts were then compared against the experimental NMR spectra. For both 1 and 2, the 13C NMR shifts could not readily distinguish the two epimers. However, the computed 1H chemical shifts for the S epimer of each compound was significantly in better agreement with the experimental values; the mean average deviation for the S epimer of 2 is 0.08 ppm but 0.36ppm for the R epimer. As a check of these results, DP4 analysis3 (see this post) of 2 indicated a 100% probability for the S epimer using only the proton chemical shifts or with the combination of proton and carbon data.
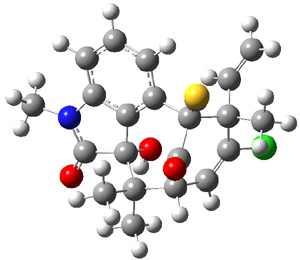 1 |
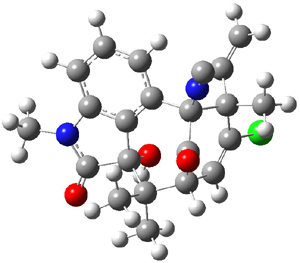 2 |
Figure 1. B3LYP/6-31+G(d,p) optimized geometries of the
lowest energy conformations of 1 and 2.
References
(1) Quasdorf, K. W.; Huters, A. D.; Lodewyk, M. W.; Tantillo, D. J.; Garg, N. K., "Total Synthesis of Oxidized Welwitindolinones and (-)-N-Methylwelwitindolinone C Isonitrile," J. Am. Chem. Soc. 2011, 134, 1396-1399, DOI: 10.1021/ja210837b
(2) Jain, R.; Bally, T.; Rablen, P. R., "Calculating Accurate Proton Chemical Shifts of Organic Molecules with Density Functional Methods and Modest Basis Sets," J. Org. Chem. 2009, 74, 4017-4023, DOI: 10.1021/jo900482q.
(3) Smith, S. G.; Goodman, J. M., "Assigning Stereochemistry to Single Diastereoisomers by GIAO NMR Calculation: The DP4 Probability," J. Am. Chem. Soc. 2010, 132, 12946-12959, DOI: 10.1021/ja105035r
InChIs
1: InChI=1/C22H21ClN2O3S/c1-6-20(4)15(23)10-13-17(26)21(20,24-11-29)12-8-7-9-14-16(12)22(28,19(13,2)3)18(27)25(14)5/h6-10,13,28H,1H2,2-5H3/t13-,20+,21+,22-/m0/s1
InChIKey=VDNAFECSPBXWIH-WOHBTAIZBQ
2: InChI=1/C22H21ClN2O3/c1-7-20(4)15(23)11-13-17(26)21(20,24-5)12-9-8-10-14-16(12)22(28,19(13,2)3)18(27)25(14)6/h7-11,13,28H,1H2,2-4,6H3/t13-,20+,21+,22-/m0/s1
InChIKey=GFNPBZSGZFQTJA-WOHBTAIZBX

Alan Shusterman responded on 02 Apr 2012 at 3:57 pm #
The DP4 method has to be taken with a grain of salt at this point in time.
First, I was unable to reproduce the list of (13C) “scaled shifts” for compd 15A, Nankakurine A in Smith and Goodman 2010, Table 2, *using the formula supplied by Smith and Goodman*. I communicated these results to Prof. Goodman in early February 2012, but the issue remains unresolved. Until my error, if it exists, is identified, I have to assume that the error lies in the published procedure and the procedure is *currently* unusable as published.
Second, DP4 method relies on several questionable assumptions about the distributions of errors, the parameters associated with this distribution, and the calculation of “probabilities” from this distribution. Therefore, one use caution in how one refers to the numerical values that DP4 assigns to probabilities. When DP4 assigns “100% probability” to a structure assignment, this does not necessarily guarantee certainty. It just means that the DP4 probability (and not even necessarily the true probability) for this structure assignment is higher than one with a lower DP4 probability value.