The di-π-methane photorearrangement has been known for many years, first studied by Zimmerman.1,2 The triplet photorearrangement gives an interesting rearranged product; and the mechanism of this photorearrangement of 1 into 2 has now been examined by the Houk group using computational techniques, including trajectory analysis. The proposed mechanism is that excitation to the triplet state 1* is followed by rearrangement to the triplet intermediate INT1* which then rearranges to the triplet INT2*. Intersystem crossing then leads to the singlet product 2.
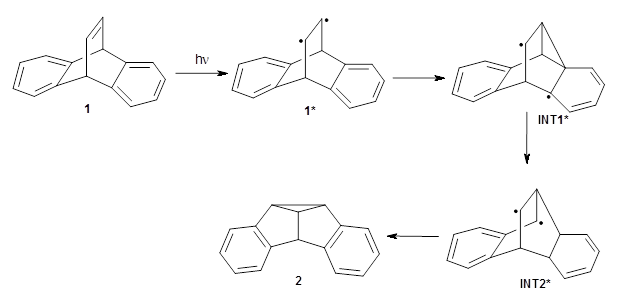
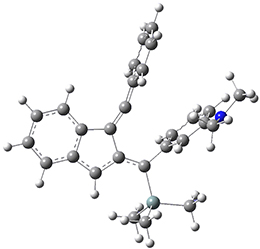
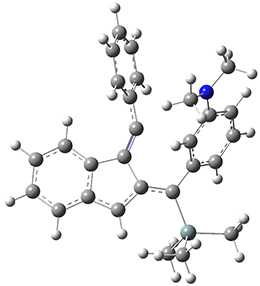
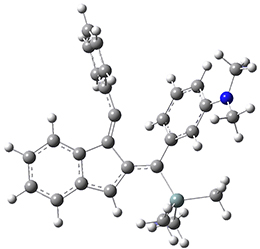
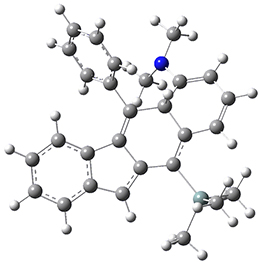
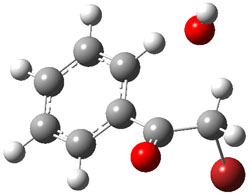
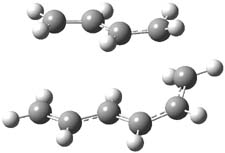
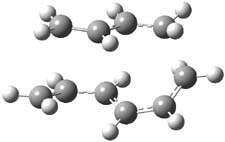
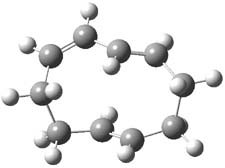
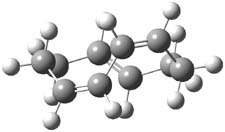
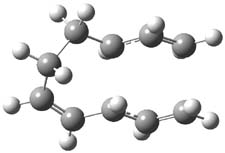
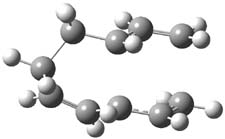
The PES for this rearrangement was explored3 at CASMP2(10,10)/6-31G(d)//CASSCF(10,10)/6-31G(d), with geometries and relative energies shown in Figure 1, as well as at (U)M06-2x/6-31G(d) and (U)B3LYP/6-31G(d); they all give qualitatively the same result. The first TS is the rate limiting step, and the second TS lies only 1-2 kcal mol-1 above the intermediate INT1. So, the reaction appears to be two steps, but with such a low barrier for the second step, dynamic effects might be important as trajectories might cross INT1* and go over TS2* without residing in the intermediate well for any appreciable time – a seemingly one step reaction. Note than no TS for directly traversing from 1* to INT2* was found.
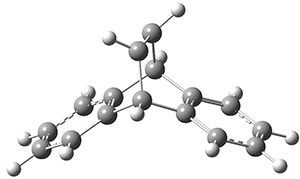
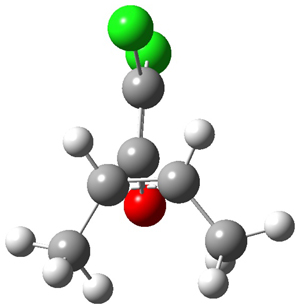
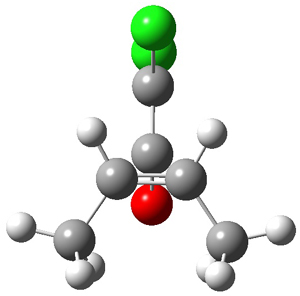
1* |
|
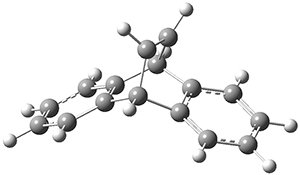
TS1* |
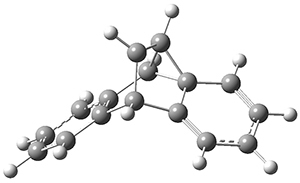
INT1* |
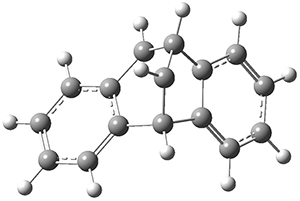
TS2* |
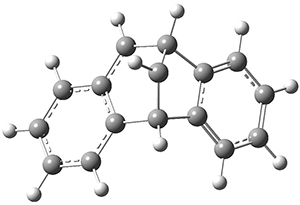
INT2* |
Figure 1. CASSCF(10,10)/6-31G(d) geometries and CASMP2 energies in kcal mol-1.
Now in a follow-up study, Houk and co-workers4 performed trajectories analysis on the M06-2x/6-31G(d) PES. A total of 256 trajectories were initiated at TS1* and 241 ended at INT2* within 1500fs. Of these, 24 trajectories resided for less than 60fs within the region of INT1, a time less than a C-C vibration. Furthermore, the lifetime of INT1 that is predicted by RRKM is much longer (about 500fs) than what is observed in the trajectories (about 200 fs). Thus, there is significant dynamic effects in this excited state rearrangement, though INT1 is always sampled.
References
(1) Zimmerman, H. E.; Grunewald, G. L. "The Chemistry of Barrelene. III. A Unique Photoisomerization to Semibullvalene," J. Am. Chem. Soc. 1966, 88, 183-184, DOI: 10.1021/ja00953a045.
(2) Zimmerman, H. E.; Binkley, R. W.; Givens, R. S.; Sherwin, M. A. "Mechanistic organic photochemistry. XXIV. The mechanism of the conversion of barrelene to semibullvalene. A general photochemical process," J. Am. Chem. Soc. 1967, 89, 3932-3933, DOI: 10.1021/ja00991a064.
(3) Matute, R. A.; Houk, K. N. "The Triplet Surface of the Zimmerman Di-π-Methane Rearrangement of Dibenzobarrelene," Angew. Chem. Int. Ed. 2012, 51, 13097-13100, DOI: 10.1002/anie.201208002.
(4) Jiménez-Osés, G.; Liu, P.; Matute, R. A.; Houk, K. N. "Competition Between Concerted and Stepwise Dynamics in the Triplet Di-π-Methane Rearrangement," Angew. Chem. Int. Ed. 2014,
53, 8664-8667, DOI: 10.1002/anie.201310237.
InChIs
1: InChI=1S/C16H12/c1-2-6-12-11(5-1)15-9-10-16(12)14-8-4-3-7-13(14)15/h1-10,15-16H
InChIKey=VWDKVBGOVYWYFZ-UHFFFAOYSA-N
2: InChI=1S/C16H12/c1-3-7-11-9(5-1)13-10-6-2-4-8-12(10)15-14(11)16(13)15/h1-8,13-16H
InChIKey=RATAQXOLJVRERC-UHFFFAOYSA-N