Once again into the breach – how much strain can an aromatic species withstand and remain aromatic? Cyranski, Bodwell and Schleyer employ the [n](2,7)pyrenophanes 1 to explore this question.1 As the tethering bridge gets shorter, the pyrene framework must pucker, presumably reducing its aromatic character. Systematic shrinking allows one to examine the loss of aromaticity as defined by aromatic stabilization energy (ASE), magnetic susceptibility exaltation (Λ) and NICS, among other measures.
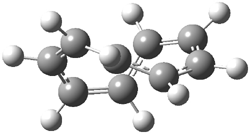
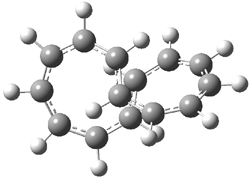
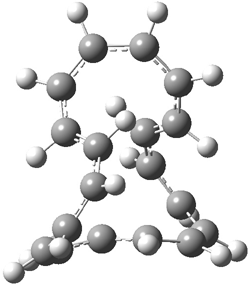
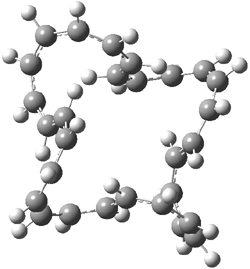
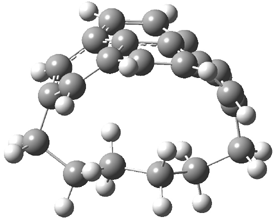
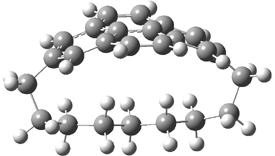
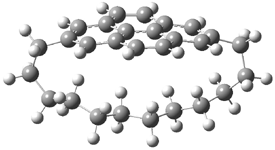
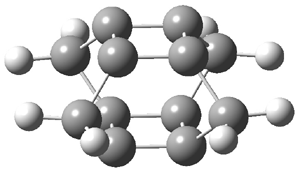
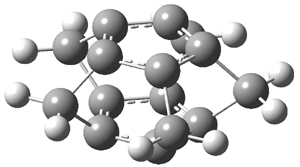
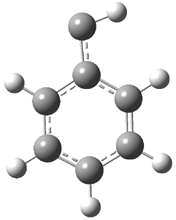
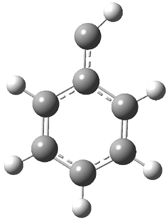
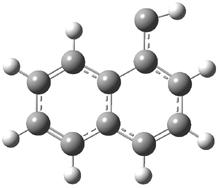
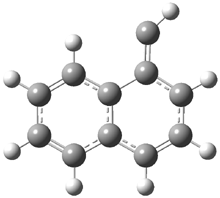
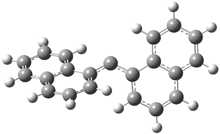
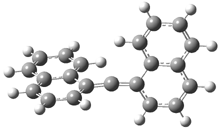
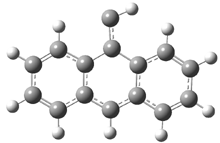
They examined the series of pyrenophanes where the tethering chain has 6 to 12 carbon atoms. I have shown the structures of three of these compounds in Figure 1. The bend angle α is defined as the angle made between the outside ring plane and the horizon. Relative ASE is computed using Reaction 1, which cleverly avoids the complication of exactly (a) what is the ASE of pyrene itself and (b) what is the strain energy in these compounds.
Figure 1. B3LYP/6-311G** optimized geometries of 1a, 1d, and 1g.1
Reaction 1
The results of the computations for this series of pyrenophanes is given in Table 1. The bending angle smoothly increases with decreasing length of the tether. The ASE decreases in the same manner. The ASE correlates quite well with the bending angle, as does the relative magnetic susceptibility exaltation. The NICS(1) values become less negative with decreasing tether length.
Table 1. Computed values for the pyrenophanes.
|
|
Compound
|
αa
|
ΔASEb
|
Rel. Λc
|
NICS(1)d
|
|
|
6(2,7)pyrenophane 1a
|
39.7
|
-15.8
|
18.8
|
-7.8, -4.1
|
|
7(2,7)pyrenophane 1b
|
32.7
|
-12.1
|
17.5
|
-8.7, -4.5
|
|
8(2,7)pyrenophane 1c
|
26.5
|
-10.6
|
14.3
|
-9.6, -5.2
|
|
9(2,7)pyrenophane 1d
|
21.3
|
-7.5
|
11.3
|
-10.6, -5.5
|
|
10(2,7)pyrenophane 1e
|
15.9
|
-6.2
|
9.5
|
-11.3, -6.2
|
|
11(2,7)pyrenophane 1f
|
11.0
|
-3.4
|
7.0
|
-12.0, -6.4
|
|
12(2,7)pyrenophane 1g
|
7.2
|
-3.1
|
6.3
|
-12.6, -7.0
|
|
pyrene
|
0.0
|
0.0
|
0.0
|
-13.9, -7.8
|
ain degrees.bin kcal mol-1, from Reaction 1.
cin cgs.ppm. din ppm, for the outer and inner rings.
|
All of these trends are consistent with reduced aromaticity with increased out-of-plane distortion of the pyrene framework. What may be surprising is the relatively small loss of aromaticity in this sequence. Even though the bend angle is as large as almost 40°, the loss of ASE is only 16 kcal mol-1, only about a quarter of the ASE of pyrene itself. Apparently, aromatic systems are fairly robust!
References
(1) Dobrowolski, M. A.; Cyranski, M. K.; Merner, B. L.; Bodwell, G. J.; Wu, J. I.; Schleyer, P. v. R.,
"Interplay of π-Electron Delocalization and Strain in [n](2,7)Pyrenophanes," J. Org. Chem., 2008, 73, 8001-8009, DOI: 10.1021/jo8014159
InChIs
1a: InChI=1/C22H20/c1-2-4-6-16-13-19-9-7-17-11-15(5-3-1)12-18-8-10-20(14-16)22(19)21(17)18/h7-14H,1-6H2
InChIKey=SJCYSWGQWCIONQ-UHFFFAOYAF
1b: InChI=1/C23H22/c1-2-4-6-16-12-18-8-10-20-14-17(7-5-3-1)15-21-11-9-19(13-16)22(18)23(20)21/h8-15H,1-7H2
InChIKey=VHVKAELFYUXZEM-UHFFFAOYAW
1c: InChI=1/C24H24/c1-2-4-6-8-18-15-21-11-9-19-13-17(7-5-3-1)14-20-10-12-22(16-18)24(21)23(19)20/h9-16H,1-8H2
InChIKey=HXPWDTNIUQNKLV-UHFFFAOYAQ
1d: InChI=1/C25H26/c1-2-4-6-8-18-14-20-10-12-22-16-19(9-7-5-3-1)17-23-13-11-21(15-18)24(20)25(22)23/h10-17H,1-9H2
InChIKey=DWYMZJZWMFVOIR-UHFFFAOYAM
1e: InChI=1/C26H28/c1-2-4-6-8-10-20-17-23-13-11-21-15-19(9-7-5-3-1)16-22-12-14-24(18-20)26(23)25(21)22/h11-18H,1-10H2
InChIKey=PZBADGOJPAEUIK-UHFFFAOYAZ
1f: InChI=1/C27H30/c1-2-4-6-8-10-20-16-22-12-14-24-18-21(11-9-7-5-3-1)19-25-15-13-23(17-20)26(22)27(24)25/h12-19H,1-11H2
InChIKey=YVZIXELCLJHDLW-UHFFFAOYAO
1g: InChI=1/C28H32/c1-2-4-6-8-10-12-22-19-25-15-13-23-17-21(11-9-7-5-3-1)18-24-14-16-26(20-22)28(25)27(23)24/h13-20H,1-12H2
InChIKey=QDAMLTATWKFTFB-UHFFFAOYAF
Pyrene: InChI=1/C16H10/c1-3-11-7-9-13-5-2-6-14-10-8-12(4-1)15(11)16(13)14/h1-10H
InChIKey=BBEAQIROQSPTKN-UHFFFAOYAB
4,9-dimethylenepyrene: InChI=1/C18H12/c1-11-9-13-5-4-8-16-12(2)10-14-6-3-7-15(11)17(14)18(13)16/h3-10H,1-2H2
InChIKey=XAAPFSHIUHWWCM-UHFFFAOYAM