Singleton has again found a great example of a simple reaction that displays unmistakable non-statistical behavior.1 The hydroboration of terminal alkenes proceeds with selectivity, preferentially giving the anti-Markovnikov product. The explanation for this selectivity is given in all entry-level organic textbooks – who would think that such a simple reaction could in fact be extraordinarily complex?
Reaction 1, designed to minimize the role of hydroboration involving higher order boron-hydrides (RBH2 and R2BH), the ratio of anti-Markovnikov to Markovinkov product is 90:10. Assuming that this ratio derives from the difference in the transition state energies leading to the two products, using transition state theory gives an estimate of the energy difference of the two activation barriers of 1.1 to 1.3 kcal mol-1.
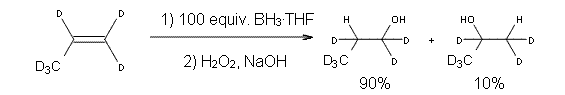
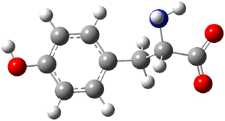
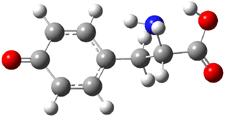
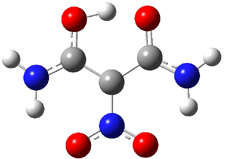
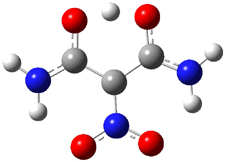
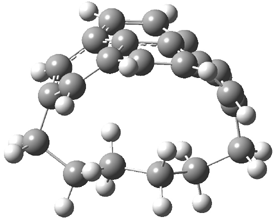
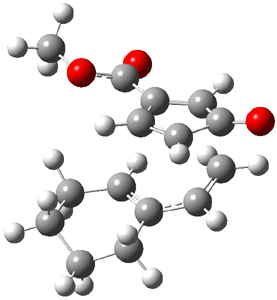
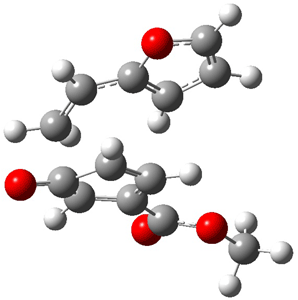
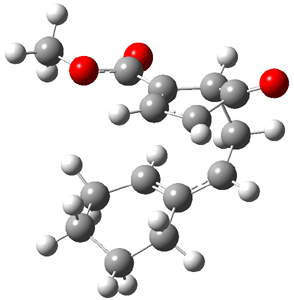
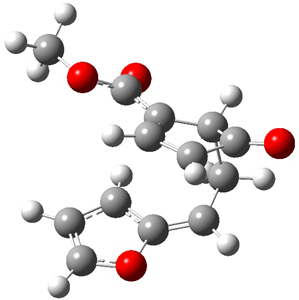
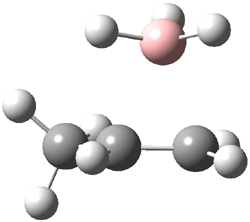
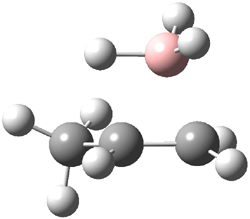
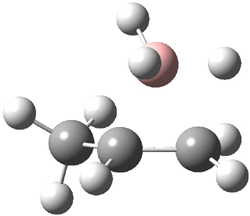
The CCSD(T)/aug-cc-pVDZ optimized structures of the precomplex between BH3 and propene 1, along with the anti-Markovnikov transition state 2 and the Markovnikov transition state 3 are shown in Figure 2. The CCSD(T) energy extrapolated for infinite basis sets and corrected for enthalpy indicate that the difference between 2 and 3 is 2.5 kcal mol-1. Therefore, transiitn state theory using this energy difference predicts a much greater selectivity of the anti-Markovnikov product, of about 99:1, than is observed.
 1 |
|
 2 |
 3 |
Figure 1. CCSD(T)/aug-cc-pVDZ optimized geometries of 1-3.1
In the gas phase, formation of the precomplex is exothermic and enthalpically barrierless. (A free energy barrier for forming the complex exists in the gas phase.) When a single THF molecule is included in the computations, the precomplex is formed after passing through a barrier much higher than the energy difference between 1 and either of the two transition states 2 or 3. (2 is only 0.8 kcal mol-1 above 1 in terms of free energy.) So, Singleton speculated that there would be little residence time within the basin associated with 1 and the reaction might express non-statistical behavior.
Classical trajectories were computed. When trajectories were started at the precomplex 1, only 1% led to the Markovnikov product, consistent with transition state theory, but inconsistent with experiment. When trajectories were initiated at the free energy transition state for formation of the complex (either with our without a single complexed THF), 10% of the trajectories ended up at the Markovnikov product, as Singleton put it “fitting strikingly well with experiment”!
Hydroboration does not follow the textbook mechanism which relies on transition state theory. Rather, the reaction is under dynamic control. This new picture is in fact much more consistent with other experimental observations, like little change in selectivity with varying alkene substitution2 and the very small H/D isotope effect of 1.18.3 Singleton adds another interesting experimental fact that does not jibe with the classical mechanism: the selectivity is little affect by temperature, showing 10% Markovnikov product at 21 °C and 11.2% Markovnikov product at 70 °C. Dynamic effect rears its ugly complication again!
References
(1) Oyola, Y.; Singleton, D. A., “Dynamics and the Failure of Transition State Theory in Alkene Hydroboration,” J. Am. Chem. Soc. 2009, 131, 3130-3131, DOI: 10.1021/ja807666d.
(2) Brown, H. C.; Moerikofer, A. W., “Hydroboration. XV. The Influence of Structure on the Relative Rates of Hydroboration of Representative Unsaturated Hydrocarbons with Diborane and with Bis-(3-methyl-2-butyl)-borane,” J. Am. Chem. Soc. 1963, 85, 2063-2065, DOI: 10.1021/ja00897a008.
(3) Pasto, D. J.; Lepeska, B.; Cheng, T. C., “Transfer reactions involving boron. XXIV. Measurement of the kinetics and activation parameters for the hydroboration of tetramethylethylene and measurement of isotope effects in the hydroboration of alkenes,” J. Am. Chem. Soc. 1972, 94, 6083-6090, DOI: 10.1021/ja00772a024.