A growing area for the application of computational chemistry is in the structural identification of compounds. In the book, I discussed the use of computed IR spectra to identify intermediates in the photolysis of phenyl nitrene and carbine and the benzynes. In previous blogs, I have written posts (here, here and here) about recent use of computed NMR spectra to discern the structure of new natural products. With this post I discus the use of computed optical activity to determine the absolute configuration of molecules.
Rosini and co-workers have examined a number of oxygenated cyclohexene epoxides.to explore the use of TDDFT computed optical activity as a means for determining absolute configuration.1 In chapter 1.6.3, I discuss the work of the Gaussian team on benchmarking optical rotation and ORD. They find that B3LYP/aug-cc-pVDZ computed optical activities are in quite reasonable agreement with experiment.2-4 In this work, Rosini explores using a smaller basis set (6-31G(d)), the role of solvent, and also if computed spectra can be used to assess the absolute configuration of new molecules.
They first benchmark the B3LYP/6-31G(d) computed optical activities for a number of related cyclohexene epoxides against B3LYP/aug-cc-pVDZ and experimental values. I will begin by discussing two of their examples: (+)-chaloxone 1 (PubChem)
and (+)-epoxydon 2
(PubChem).

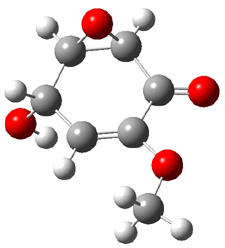
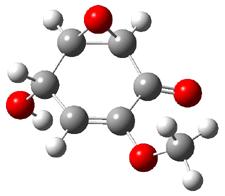
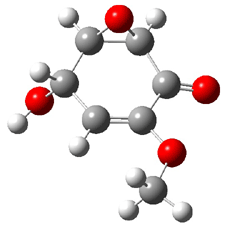
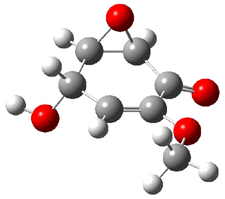
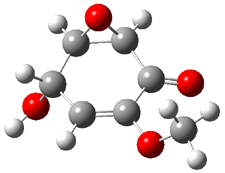
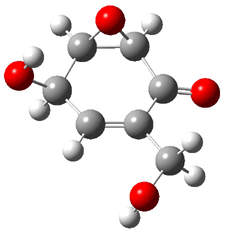
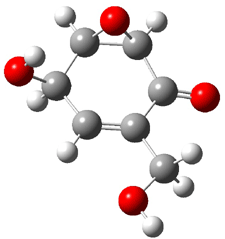
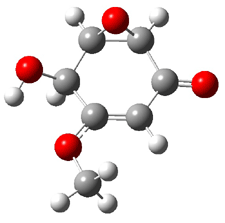
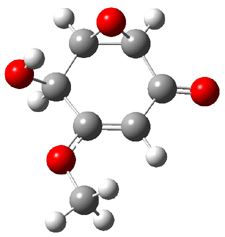
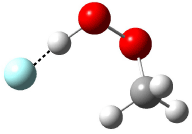
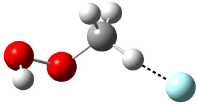
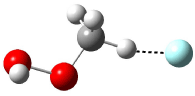
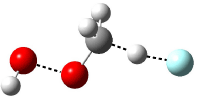
Five conformations of 1 were optimized in the gas phase and then their optical activities for the sodium D line were computed using TDDFT with both the small and larger basis set. These computations were then repeated to model the effect of solvent using PCM; the solution (methanol) B3LYP/6-31G(d) structures are shown in Figure 1.
|
1a
0.0
|
1b
2.40
|
1c
0.87
|
|
1d
1.02
|
1e
3.12
|
|
Figure 1. PCM(methanol)/B3LYP/6-31G(d) optimized structures of 1. Relative free energies of each conformer in kcal/mol.1
The optical rotation at the sodium D line was then computed with TDDFT in both gas and solution phase with the smaller and larger basis set. The values were then averaged base on a Boltzmann weighting using the computed free energies of each conformer. The optical rotation for each conformer and the average values are listed in Table 1. The experimental optical rotation is +271. The authors note that while the gas phase B3LYP/6-31G(d) average value is far off the experimental value, it does predict the correct sign, and since all of the five conformers give rise to a positive rotation, any error in the energies will not affect the sign. The computed gas phase value with the larger basis set is in better agreement with experiment. However, it is still too large, but the solution values are much better. In fact, the PCM/B3LYP/aug-cc-pVDZ value is in excellent agreement with experiment.
Table 1. Computed optical activity of the conformers of 1 in gas and solution phase.
|
|
|
gas
|
solution
|
|
conformer
|
6-31G(d)
|
aug-cc-pvDZ
|
6-31G(d)
|
aug-cc-pvDZ
|
|
1a
|
+264
|
+251
|
+304
|
+308
|
|
1b
|
+723
|
+750
|
+690
|
+707
|
|
1c
|
+324
|
+309
|
+398
|
+385
|
|
1d
|
+187
|
+201
|
+246
|
+268
|
|
1e
|
+741
|
+785
|
+756
|
+769
|
|
Averagea
|
+378
|
+333
|
+318
|
+322
|
|
aBased on a Boltzmann weighting of the population of each conformation.
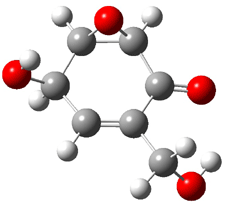
Five conformers of epoxydon 2 were also located, and the computed solution structures are shown in Figure 2. The computed optical rotations for both the gas and solution phase for these structures (and the Boltzmann weighted averages) are listed in Table 2. The experimental value for the optical rotation of 2 is +93.
|
2a
0.0
|
2b
0.32
|
2c
0.23
|
|
2d
0.22
|
2e
0.66
|
|
Figure 2. PCM(methanol)/B3LYP/6-31G(d) optimized structures of 2. Relative free energies of each conformer in kcal/mol.1
In this case, the small basis set performs very poorly. The gas phase B3LYP/6-31G(d) value
of [α]D is -16, predicting the wrong sign, let alone the wrong magnitude. Things improve with the larger basis set, which predicts a value of +57. Since conformer 2ais levorotatory and the other four are dextrorotatory, the computed relative energies are key to getting the correct prediction. This is made even more poignant with the solution results, where the PCM/B3LYP/aug-cc-pVDZ prediction is quite acceptable.
Table 2. Computed optical activity of the conformers of 2 in gas and solution phase.
|
|
gas
|
solution
|
|
conformer
|
6-31G(d)
|
aug-cc-pvDZ
|
6-31G(d)
|
aug-cc-pvDZ
|
|
2a
|
-97
|
-43
|
-85
|
-36
|
|
2b
|
+130
|
+210
|
+113
|
+166
|
|
2c
|
+14
|
+63
|
+8
|
+58
|
|
2d
|
+113
|
+119
|
+37
|
+71
|
|
2e
|
+29
|
+86
|
+19
|
+67
|
|
Averagea
|
-16
|
+57
|
+4
|
+61
|
|
aBased on a Boltzmann weighting of the population of each conformation.
Threy conclude with two examples of application of computation to assignment of structure. I discuss here the absolute configuration of (-)-sphaeropsidone 3 (PubChem).
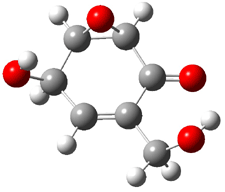
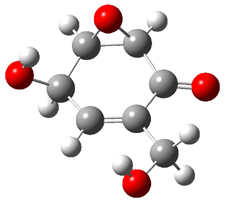
Rosini located two conformations of 3, shown in Figure 3. The computed optical rotations are listed in Table 3. The experimental value for 3 is -130. Both conformers are computed to be dextrorotatory with all computational methods. The magnitude of the computed values using the larger basis set is in nice agreement with experiment, but the sign is wrong. Rosini concludes that the absolute configuration of 3 has been misassigned.
Figure 3. PCM(methanol)/B3LYP/6-31G(d) optimized structures of 3. Relative free energies of each conformer in kcal/mol.1
Table 3. Computed optical activity of the conformers of 3 in gas and solution phase.
|
|
gas
|
solution
|
|
conformer
|
6-31G(d)
|
aug-cc-pvDZ
|
6-31G(d)
|
aug-cc-pvDZ
|
|
3a
|
+99
|
+172
|
+67
|
+135
|
|
3b
|
+54
|
+109
|
+20
|
+69
|
|
Averagea
|
+85
|
+146
|
+43
|
+101
|
|
aBased on a Boltzmann weighting of the population of each conformation.
References
(1) Mennucci, B.; Claps, M.; Evidente, A.; Rosini, C., "Absolute Configuration of Natural Cyclohexene Oxides by Time Dependent Density Functional Theory Calculation of the Optical Rotation: The Absolute Configuration of (-)-Sphaeropsidone and (-)-Episphaeropsidone Revised," J. Org. Chem. 2007, 72, 6680-6691, DOI: 10.1021/jo070806i
(2) Stephens, P. J.; Devlin, F. J.; Cheeseman, J. R.; Frisch, M. J., "Calculation of Optical Rotation Using Density Functional Theory," J. Phys. Chem. A 2001, 105, 5356-5371, DOI: 10.1021/jp0105138.
(3) Stephens, P. J.; McCann, D. M.; Cheeseman, J. R.; Frisch, M. J., "Determination of
absolute configurations of chiral molecules using ab initio time-dependent Density Functional Theory calculations of optical rotation: How reliable are absolute configurations obtained for molecules with small rotations?," Chirality 2005, 17, S52-S64, DOI: 10.1002/chir.20109.
(4) Stephens, P. J.; McCann, D. M.; Devlin, F. J.; Flood, T. C.; Butkus, E.; Stoncius,
S.; Cheeseman, J. R., "Determination of Molecular Structure Using Vibrational Circular Dichroism Spectroscopy: The Keto-lactone Product of Baeyer-Villiger Oxidation of (+)-(1R,5S)-Bicyclo[3.3.1]nonane-2,7-dione," J. Org. Chem. 2005, 70, 3903-3913, DOI: 10.1021/jo047906y.
InChI
1: InChI=1/C7H8O4/c1-10-4-2-3(8)6-7(11-6)5(4)9/h2-3,6-8H,1H3
2: InChI=1/C7H8O4/c8-2-3-1-4(9)6-7(11-6)5(3)10/h1,4,6-9H,2H2
3: InChI=1/C7H8O4/c1-10-4-2-3(8)6-7(11-6)5(4)9/h2,5-7,9H,1H3