Saelli, Nicolaou, and Bagno point out in a recent article how the determination of the structure of vannusal B might have been guided by DFT computed 13C NMR chemical shifts, had they been available.1 The original structure was proposed in 1999 as 1,2 but was ultimately settled as 2 in 2010.3
|
|
|
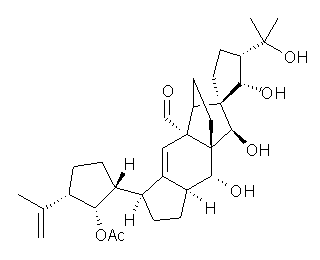
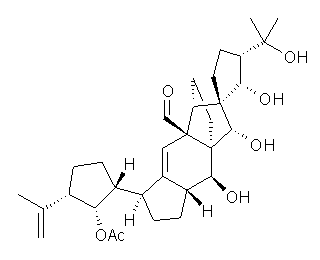
The 13C NMR chemical shifts of 1 and 2 and some other alternatives were computed at M06/pcS-2//B3LYP/6-31g(d,p), where the pcS-2 basis set4 is one proposed by Jensen for computing chemical shifts. The computed chemical shifts of 1 poorly correlate with the experimental chemical shifts of vannusal B, with a low correlation coefficient of 0.9580 and a maximum error of 16.2 ppm. On the other hand, the correlation between the computed chemical shifts of 2 with the experimental values is excellent (R2=0.9948) and a maximum error of 3.0 ppm. Comparison of computed and experimental H-H coupling constants of model compounds of the “northeast” section of the molecule verified the correct structure is 2.
References
(1) Saielli, G.; Nicolaou, K. C.; Ortiz, A.; Zhang, H.; Bagno, A., "Addressing the Stereochemistry of Complex Organic Molecules by Density Functional Theory-NMR: Vannusal B in Retrospective," J. Am. Chem. Soc., 2011, 133, 6072-6077, DOI: 10.1021/ja201108a
(2) Guella, G.; Dini, F.; Pietra, F., "Metabolites with a Novel C30 Backbone from Marine Ciliates," Angew. Chem. Int. Ed., 1999, 38, 1134-1136, DOI: 10.1002/(SICI)1521-3773(19990419)38:8<1134::AID-ANIE1134>3.0.CO;2-U
(3) Nicolaou, K. C.; Ortiz, A.; Zhang, H.; Dagneau, P.; Lanver, A.; Jennings, M. P.; Arseniyadis, S.; Faraoni, R.; Lizos, D. E., "Total Synthesis and Structural Revision of Vannusals A and B: Synthesis of the Originally Assigned Structure of Vannusal B," J. Am. Chem. Soc., 2010, 132, 7138-7152, DOI: 10.1021/ja100740t
(4) Jensen, F., "Basis Set Convergence of Nuclear Magnetic Shielding Constants Calculated by Density Functional Methods," J. Chem. Theory Comput., 2008, 4, 719-727, DOI: 10.1021/ct800013z
InChI
vannusal B (2):
InChI=1/C31H46O5/c1-16(2)18-6-7-19(17(18)3)20-8-9-21-22(20)14-29(15-32)24-11-13-31(29,25(21)33)27(35)30(24)12-10-23(26(30)34)28(4,5)36/h14-15,17-21,23-27,33-36H,1,6-13H2,2-5H3/t17-,18+,19+,20+,21-,23+,24+,25+,26?,27+,29+,30-,31-/m1/s1
InChIKey=KYOBJLKAZYUEHK-GYGUSHOLBX

Henry Rzepa responded on 25 May 2011 at 10:12 am #
For these hydroxylated rings with little unsaturation, hexacyclinol remains the key molecule. It turns out that tinkering with either the functional or the basis set changes the predictions only modestly. Thus we reduced the error in hexacyclinol itself down to a RMS of ~1ppm (DOI: 10.1021/np0705918), mostly by careful attention to solvation rather than functional or basis set. Molecule 2 above is also slightly atypical; it is very rigid conformationally. Whilst this matters less for 13C, conformation CAN affect shifts by 2-5ppm.
There is little doubt that the last five years have brought about a true sea change; the range and quality of computationally predicted spectroscopies (13C/1H/15N/19F NMR, UV-Vis (and the chiroptical equivalents, ECD), vibrational (and chiroptical versions VCD and ROA, and finally ORP and ORD (using modern polarimeters that can range from ~250nm to 880nm and beyond) mean that no synthetic chemist can nowadays afford to be without access to such computations (and they MUST have the mindset to go get this).
Steven Bachrach responded on 25 May 2011 at 11:10 am #
I completely agree with Henry’s closing statement. The most important advancement in the past 5-10 years in computational organic chemistry has been the development of reasonable computational methods for determining reasonably accurate optical (and chiro-optical) properties.
Henry Rzepa responded on 25 May 2011 at 1:02 pm #
Can I pick up on Steve’s last comment, and seek help! The chemical heritage of my department includes two iconic colours, mauveine and monastral blue. For talk given to students on art, a colleague asked me to identify the electronic origins of the colours of these two species. I tried, and failed on both of them. With mauveine, predicted &lamba;max was out by 100nm (440 instead of 540nm). With copper phthalocyanine, TD-DFT gets one band correct (610nm) but fails entirely to predict another strong one (710). I have tried lots of different models, and all fail. By way of the open science model, what does the community think?