A nice follow-up to some of my own work points out again the possible dramatic role of dynamic effects. Way back when, Jack Gilbert discovered that the reaction of cyclopentyne with alkenes gives the cyclobutene product with stereoretention (Reaction 1),1 seemingly in violation of the Woodward-Hoffmann rules.
|
|
Reaction 1 |
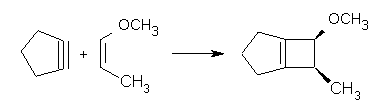
Jack and I proposed an intermediate spirocyclopropyl carbene which could then open to product, and this would follow a stereoretention path.2,3 In a subsequent paper,4 we noted that a diradical pathway is also possible, and conjectured that dynamics might account for the stereoretention – that formation of the diradical leads directly to the carbene, leaving a very short lifetime of the diradical (Scheme 1). The consequence of the short lived diradical is that there little opportunity to rotate about the C-C bond and scramble the stereochemistry.
Scheme 1
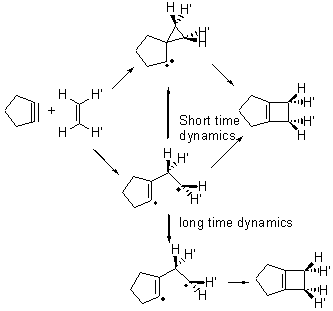
Pilling has published a MD study of this system and finds what we predicted.5 The short-time trajectories lead to stereoretention product. This is due to both passages over the TS that lead from the diradical to the product (with no scrambling) and over the TS that connects the diradical to the carbine. Longer trajectories do exhibit some stereoscrambling. Carpenter6 has argued that short time dynamics are often what one observes for potential energy surfaces like this one. Pilling also argues that in solution, with the actual alkene which bears bulky substituents that the proton (he examined the reaction of cyclopentyne with ethene), rotations will be slower, leading to formation of the carbene with stereoretention.
References
(1) Gilbert, J. C.; Baze, M. E., "Stereochemistry of [2 + 2] cycloadditions of cyclopentyne," J. Am. Chem. Soc. 2002, 106, 1885-1886, DOI: 10.1021/ja00318a081
(2) Laird, D. W.; Gilbert, J. C., "Norbornyne: A Cycloalkyne Reacting Like A Dicarbene," J. Am. Chem. Soc., 2001, 123, 6704-6705, DOI: 10.1021/ja010589h
(3) Bachrach, S. M.; Gilbert, J. C.; Laird, D. W., "DFT Study of the Cycloaddition Reactions of Strained Alkynes," J. Am. Chem. Soc., 2001, 123, 6706-6707, DOI: 10.1021/ja010590g
(4) Bachrach, S. M.; Gilbert, J. C., "The Reaction of Cyclopentyne with Ethene: Concerted vs Stepwise Mechanism?," J. Org. Chem., 2004, 69, 6357-6364, DOI: 10.1021/jo0492970
(5) Glowacki, D. R.; Marsden, S. P.; Pilling, M. J., "Significance of Nonstatistical Dynamics in Organic Reaction Mechanisms: Time-Dependent Stereoselectivity in Cyclopentyne−Alkene Cycloadditions," J. Am. Chem. Soc. 2009, 131, 13896-13897, DOI: 10.1021/ja9043054
(6) Barry, K. C., "Nonexponential decay of reactive intermediates: new challenges for spectroscopic observation, kinetic modeling and mechanistic interpretation," J. Phys. Org. Chem., 2003, 16, 858-868, DOI: 10.1002/poc.672

Henry Rzepa responded on 18 Nov 2009 at 3:54 am #
I am interested in one aspect of Steve’s original study, which found a concerted pathway for reaction of an alkyne to give a spiro-cyclopropane carbene. The formal arrow pushing has one unusual and rare feature; namely two electron pairs move away from the triple bond at the same time. In my experience, such (formal) mechanisms are quite rare (other examples I can think of is the reaction between a per-acid and an alkyne, which produces initially a ketocarbene, the dimerisation of a carbene to give an alkene, and some double [3,3] sigmatropic reactions involving alkynes). If one pauses to consider, most arrow pushing changes the bond order of any given bond by only one (i.e. zero to one, one to zero, two to one, etc etc).
I am always keen to collect further examples of such large formal bond order changes which occur during a concerted (if not synchronous) mechanistic step. Whilst I suspect that Δ2 in the bond order is likely to be the maximum for carbon bonds, there may be examples of higher involving eg metals. So if anyone knows of any and is willing to share them here … ?
I also somehow feel that we should have a name for any concerted mechanistic step involving such high bond order changes. Perhaps there already is one?
Mechanistic Ménage à trois « Henry Rzepa responded on 18 Nov 2009 at 7:17 am #
[…] Curly arrow pushing is one of the essential tools of a mechanistic chemist. Many a published article will speculate about the arrow pushing in a mechanism, although it is becoming increasingly common for these speculations to be backed up by quantitative quantum mechanical and dynamical calculations. These have the potential of exposing the underlying choreography of the electronic dance (the order in which the steps take place). The basic grammar of describing that choreography tends to be the full-headed curly arrow for closed shell systems and its half-barbed equivalent for open shell systems. An effectively unstated and hence implicit rule for closed shell systems is that only one curly arrow is used per breaking or forming bond, i.e. electrons move around bonds in pairs. So consider the following reaction (inspired by a posting on Steve Bachrach’s blog) […]
Henry Rzepa responded on 23 Nov 2009 at 11:53 am #
About 11 years ago, we suggested yet another alternative to the Woodward-Hoffmann forbidden π2s + π2s cycloaddition or the π2s + π2a allowed variation (DOI: 10.1039/a805668d). This involved a trapezoidal distortion to the geometry of the transition state which eliminated the forbidden orbital overlap, and allowed a synchronous potential surface. I wonder whether such a region of the PES has been explored for the reactions described in this blog entry? Might this model also lead to stereo-retention?