Schaefer and Allen have applied their focal point method to the question of the benzylic effect in the SN2 reaction.1 SN2 reactions are accelerated when the attack occurs at the benzylic carbon, a well-known phenomenon yet the reason for this remains unclear. The standard textbook-like argument has been that the negative charge built up in the SN2 transition state can be delocalized into the phenyl ring. However, solution phase Hammett studies are often U-shaped, indicating that both electron donating and withdrawing group accelerate the substitution reaction. (This is usually argued as indicative of a mechanism change from SN2 to SN1.)
The focal point method involves a series of very large computations where both basis set size and degree of electron correlation are systematically increased, allowing for an extrapolation to essentially infinite basis set and complete correlation energy. The energy of the transition state (relative to separated reactants) for four simple SN2 reactions evaluated with the focal point method are listed in Table 1. The barrier for the benzylic substitutions is lower than for the methyl cases, indicative of the benzylic effect.
Table 1. Energy (kcal mol-1) of the transition state relative to reactants.1
|
|
|||
|
|
Ea |
Ea |
|
|
F– + CH3F |
-0.81 |
-2.42 |
|
|
F– + PhCH2F |
-4.63 |
-5.11 |
|
|
Cl– + CH3Cl |
+1.85 |
-1.31 |
|
|
Cl– + PhCH2Cl |
+0.24 |
-2.11 |
|
|
|
|||
To answer the question of why the benzylic substitution reactions are faster, they examined the charge distribution evaluated at B3LYP/DZP++. As seen in Table 1, this method does not accurately reproduce the activation barriers, but the errors are not terrible, and the trends are correct.
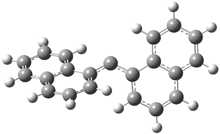
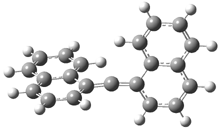
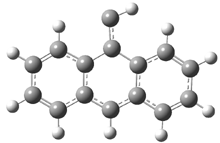
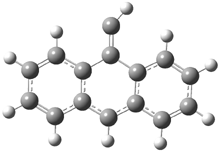
In Figure 1 are the geometries of the transition states for the reaction of fluoride with methylflouride or benzylfluoride. The NBO atomic charges show that the phenyl ring acquired very little negative charge at the transition state. Rather, the electric potential at the carbon under attack is much more revealing. The potential is significantly more positive for the benzylic carbon than the methyl carbon in both the reactant and transition states.
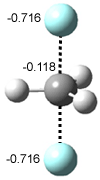 |
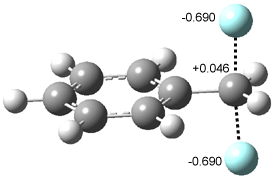 |
|
VC = -405.156 V |
VC = -404.379 V |
Figure 1. MP2/DZP++ transition states for the reaction of fluoride with methylfluoride and benzylflouride. NBO charges on F and C and the electrostatic potential in Volts.1
They next examined the reaction of fluoride with a series of para-substituted benzylfluorides. The relation between the Hammet σ constants and the activation energy is fair (r = 0.971). But the relation between the electrostatic potential at the benzylic carbon (in either the reactant or transition state) with the activation energy is excellent (r = 0.994 or 0.998). Thus, they argue that it is the increased electrostatic potential at the benzylic carbon that accounts for the increased rate of the SN2 reaction.
References
(1) Galabov, B.; Nikolova, V.; Wilke, J. J.; Schaefer III, H. F.; Allen, W. D., "Origin of the SN2 Benzylic Effect," J. Am. Chem. Soc., 2008, 130, 9887-9896, DOI: 10.1021/ja802246y.