With the proliferation of density functionals, selecting the functional to use in your particular application requires some care. That is why there have been quite a number of benchmark studies (see these posts for some examples). Yu and Karton have now added to our benchmark catalog with a study of π-conjugation.1
They looked at a set of 60 reactions which involve a reactant with π-conjugation and a product which lacks conjugation. A few examples, showing examples involving linear and cyclic systems, are shown in Scheme 1.
Scheme 1.
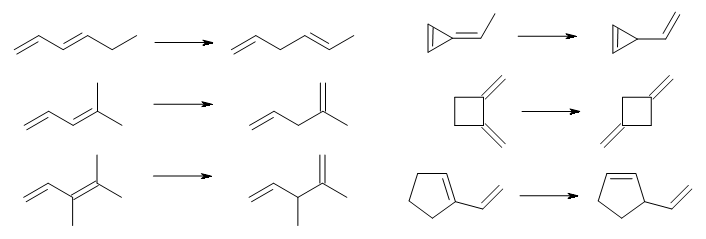
The reaction energies were evaluated at W2-F12, which should have an error of a fraction of a kcal mol-1. Three of the reactions can be compared with experimental values, and difference in the experimental and computed values are well within the error bars of the experiment. It is too bad that the authors did not also examine 1,3-cyclohexadiene → 1,4-cyclohexadiene, a reaction that is both of broader interest than many of the ones included in the test set and can also be compared with experiment.
These 60 reactions were then evaluated with a slew of functionals from every rung of Jacob’s ladder. The highlights of this benchmark study are that most GGA and meta-GGA and hybrid functionals (like B3LYP) have errors that exceed chemical accuracy (about 1 kcal mol-1). However, the range-separated functionals give very good energies, including ωB97X-D. The best results are provided with double hybrid functionals. Lastly, the D3 dispersion correction does generally improve energies by 10-20%. On the wavefunction side, SCS-MPs gives excellent results, and may be one of the best choices when considering computational resources.
References
(1) Yu, L.-J.; Karton, A. "Assessment of theoretical procedures for a diverse set of isomerization reactions involving double-bond migration in conjugated dienes," Chem. Phys. 2014, 441, 166-177, DOI: 10.1016/j.chemphys.2014.07.015.

Amir Karton responded on 19 Jan 2015 at 6:39 am #
Thank you for this highlight and the suggestion to examine the 1,3-cyclohexadiene → 1,4-cyclohexadiene isomerization. We ran the W2-F12 calculations and we find that this reaction is nearly thermoneutral at the W2-F12 level. Namely, the W2-F12 CCSD(T)/CBS isomerization energy at 298 K is -0.057 kcal/mol (for comparison the experimental value taken from the NIST thermochemical database is +0.041 kcal/mol). Here is the breakdown of the W2-F12 components for this isomerization reaction (in kcal/mol):
∆Escf = -1.801
∆Eccsd = 1.180
∆E(T) = 0.662
∆Ecv = -0.041
∆Erel = 0.003
∆Edboc = 0.002
∆Eisomer,elec = 0.005
∆Hisomer,0K = -0.107
∆Hisomer,298K = -0.057
For evaluation of the DFT functionals we obtain a nonrelativistic, all-electron, vibrationless CCSD(T)/CBS energy of 0.0001 kcal/mol at the W2-F12 level. The DFT functionals that were considered in the paper perform reasonably well for this reaction, with isomerization energies ranging between -1.10 (LC-wPBE) and +0.74 (B2K-PLYP). We note that 76% of the fuctionals give isomerization energies (in absolute value) ≤ 0.5 kcal/mol, and 11 functionals give isomerization energies (in absolute value) ≤ 0.1 kcal/mol. Specifically, -0.10 (B3LYP), -0.10 (M05-2X), -0.07 (M06-HF), -0.07 (B98), -0.02 (B97-1), +0.02 (M11-L), +0.02 (t-HCTHh), +0.05 (DSD-PBEP86), +0.05 (t-HCTH), +0.07 (B3PW91-D3), and +0.10 (TPSSh) kcal/mol.
Steven Bachrach responded on 19 Jan 2015 at 10:33 am #
Thanks Amir for this quick addition to your study! Remarkably B3LYP performs so well!