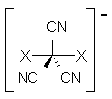
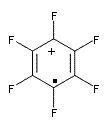
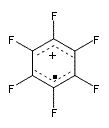
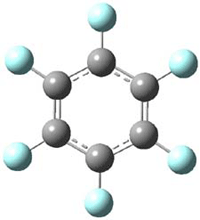
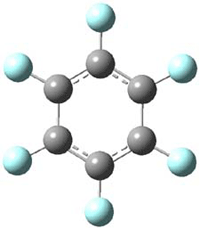
Though not quite germane to this blog (Computational Organic Chemistry), the recent commentary by Rzepa1 does deserve mention. Henry takes on, in a nice breezy style (note the title: The importance of being bonded), the nature of bonding in 1, which initially was thought to be of structure 1a but subsequent x-ray structural analysis suggested the presence of an S-S bond, i.e. 1b. Schleyer has applied NICS analysis to suggest that the compound is bishomoaromatic.2 Henry utilizes AIM and ELF analysis to discuss the nature of the bonding, including the possibility of H…H interaction between the methyl groups and trishomoaromatic character. What I liked about the article is that Henry rightly makes the case that exploration of the notion of “bonding” can be quite opaque and often leads to stretching the models we commonly employ. Well worth the read!

References
(1) Rzepa, H. S., "The importance of being bonded," Nat. Chem., 2009, 1, 510-512, DOI: 10.1038/nchem.373.
(2) Zhang, Q.; Yue, S.; Lu, X.; Chen, Z.; Huang, R.; Zheng, L.; Schleyer, P. v. R., "Homoconjugation/Homoaromaticity in Main Group Inorganic Molecules," J. Am. Chem. Soc., 2009, 131, 9789-9799, DOI: 10.1021/ja9029285