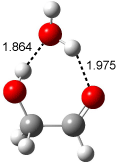
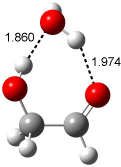
Bertran and Wipf have examined the role of solvent organization about a chiral molecule in producing the optical activity.1 They generated 1000 configurations of benzene arrayed about methyloxirane from a Monte Carlo simulation. Each configuration was then constructed by keeping every benzene molecules within 0.5 nm from the center-of-mass of methyloxirane, usually 8-10 solvent molecules. The optical rotation was then computed at four wavelengths using TDDFT at BP86/SVP. (The authors note that though the Gaussian group recommends B3LYP/aug-ccpVDZ,2-4 using the non-hybrid functional allows the use of resolution-of–the-identity5 techniques that make the computations about six orders of magnitude faster – of critical importance given the size of the clusters and the sheer number of them!) Optical rotation is then obtained by averaging over the ensemble.
The computed optical rotations disagree with the experiment by about 50% in magnitude but have the correct sign across the four different wavelengths. Use of the COSMO model (implicit solvent) provides the wrong sign at short wavelengths. But perhaps most interesting is that the computed optical activity of the solvent molecules in the configuration about the solute, but without including methyloxirane, is nearly identical to that of the whole cluster! In other words, the optical activity is due to the dissymmetric distribution of the solvent molecules about the chiral molecule, not the chiral molecule itself! It is the imprint of the chiral molecule on the solvent ordering that accounts for nearly all of the optical activity.
References
(1) Mukhopadhyay, P.; Zuber, G.; Wipf, P.; Beratan, D. N., "Contribution of a Solute’s
Chiral Solvent Imprint to Optical Rotation," Angew. Chem. Int. Ed. 2007,
46, 6450-6452, DOI: 10.1002/anie.200702273
(2) Stephens, P. J.; McCann, D. M.; Cheeseman, J. R.; Frisch, M. J., "Determination of
absolute configurations of chiral molecules using ab initio time-dependent Density Functional Theory calculations of optical rotation: How reliable are absolute configurations obtained for molecules with small rotations?," Chirality 2005, 17, S52-S64, DOI: 10.1002/chir.20109.
(3) Stephens, P. J.; Devlin, F. J.; Cheeseman, J. R.; Frisch, M. J., "Calculation of Optical Rotation Using Density Functional Theory," J. Phys. Chem. A 2001, 105, 5356-5371, DOI: 10.1021/jp0105138.
(4) Stephens, P. J.; McCann, D. M.; Devlin, F. J.; Flood, T. C.; Butkus, E.; Stoncius,
S.; Cheeseman, J. R., "Determination of Molecular Structure Using Vibrational Circular Dichroism Spectroscopy: The Keto-lactone Product of Baeyer-Villiger Oxidation of (+)-(1R,5S)-Bicyclo[3.3.1]nonane-2,7-dione," J. Org. Chem. 2005, 70, 3903-3913, DOI: 10.1021/jo047906y.
(5) Eichkorn, K.; Treutler, O.; Ohm, H.; Haser, M.; Ahlrichs, R., "Auxiliary Basis Sets to Approximate Coulomb Potentials," Chem. Phys. Lett. 1995, 240, 283-289, DOI: 10.1016/0009-2614(95)00621-A.