Cramer and Hoye have applied DFT computations to the predictions of both protons and carbon NMR chemical shifts in penam β-lactams1 using the procedure previously described in my blog post Predicting NMR chemical shifts. They examined the compounds 1-8 by optimizing low energy conformers at B3LYP/6-31G(d) with IEFPCM (solvent=chloroform). The chemical shifts were then computed using these geometries with the larger 6-311+G(2d,p) basis set and four different functionals: B3LYP, PBE1 and the two specific functionals designed to produce proton and carbon chemical shifts: WP04 and WC04.
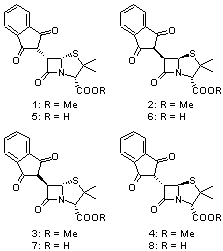
A number of interesting results are reported. First, all three functionals do a fine job in predicting the proton chemical shifts of 1-8, with WP04 slightly better than the other two.On the other hand, all three methods fail to predict the carbon chemical shifts of 1-3, though B3LYP and PBE1 do correctly identify 5-8. The failure of WC04 is surprising, especially since dimethyl disulfide was used in the training set. They also noted that WP04 using just the minimum energy conformation (as opposed to a Boltzmann averaged chemical shift sampled from many low energy conformers) did correctly identify lactams 1-4. This is helped by the fact that the lowest energy conformer constituted anywhere form 37% to 68% of the energy-weighted population.
References
(1) Wiitala, K. W.; Cramer, C. J.; Hoye, T. R., “Comparison of various density functional methods for distinguishing stereoisomers based on computed 1H or 13C NMR chemical shifts using diastereomeric penam ?-lactams as a test set,” Mag. Reson. Chem., 2007, 45, 819-829, DOI: 10.1002/mrc.2045.
InChIs
1: InChI=1/C18H17NO5S/c1-18(2)14(17(23)24-3)19-15(22)11(16(19)25-18)10-12(20)8-6-4-5-7-9(8)13(10)21/h4-7,10-11,14,16H,1-3H3/t11-,14+,16+/m0/s1
5: InChI=1/C17H15NO5S/c1-17(2)13(16(22)23)18-14(21)10(15(18)24-17)9-11(19)7-5-3-4-6-8(7)12(9)20/h3-6,9-10,13,15H,1-2H3,(H,22,23)/t10-,13+,15+/m0/s1
