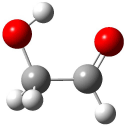
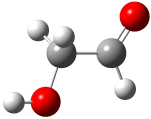
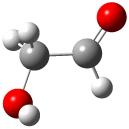
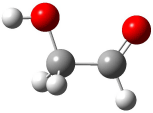
A Stable Bis-allyl Intermediate on the Cope PES
As discussed in Chapter 3.2, the prototypical Cope rearrangement (the degenerate rearrangement of 1,5-hexadiene 1) is understood to proceed through a single concerted transition state. The concerted transition state 2 is described by three resonance structures (Scheme 1), and this allows for understanding the chameleonic nature of the substituted Cope rearrangement. For example, radical stabilizing groups at the 2 and 5 positions would favor the cyclohexyl-diyl structure.
Scheme 1

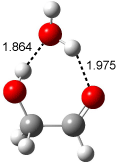
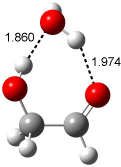
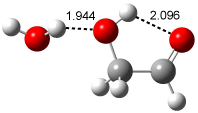
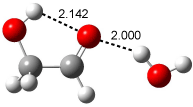
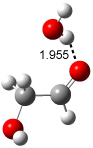
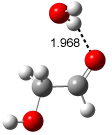
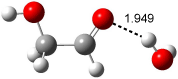
Schreiner computed the reaction path at BD(T)/cc-pVDZ//BLYP/6-31G* for 64 different variations of the Cope rearrangement.1 A representative sampling from these is presented in Figure 1. The Cope rearrangements are found to fall into one of three categories. The first, called type 1, are concerted rearrangements. Type 2 rearrangements have two competing pathways: either through a concerted transition state or a diradical intermediate. The last group, type 3, comprises nonconcerted reactions with a cyclohexyl-diyl intermediate. Schreiner generalizes the results to the following rule: a nonconcerted reaction takes place when biradical intermediates are stabilized either by allyl or aromatic resonance.
 |
|
Figure 1. Examples of the three type of Cope rearrangements. Relative energies, in kcal mol-1, were computed at BD(T)/cc-pVDZ//BLYP/6-31G*.1
|
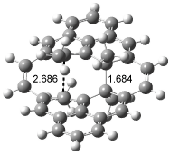
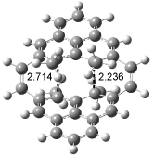
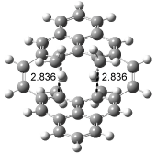
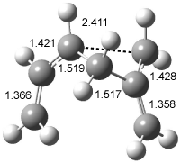
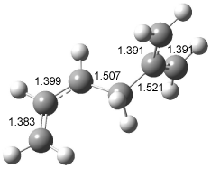
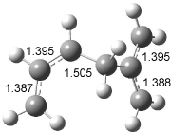
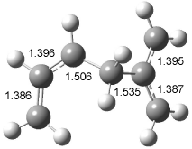
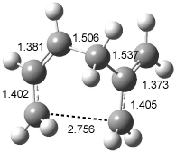
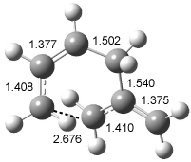
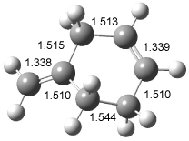
Interestingly, Schreiner’s study identified reactions where the diyl is a stable intermediate, but he identified no case where the other extreme – two allyl radicals – appeared as a stable intermediate. Kertesz, in 2006, discovered just such an example with the Cope rearrangement of 3.2 Using B3LYP and BPW91 computations with two different basis sets, he identified the stable diradical 5. This structure, shown in Figure 2, clearly has very long distances – 2.836 Å – separating the ends of the two “allylic” components. A true transition state 4 connects the reactant 3 with the intermediate 5 (see Figure 2). The activation energy is 6.3 kcal mol-1 and the intermediate 5 lies 3.3 kcal mol-1 above 3.
 |
|
|
|
|
| 3 |
4 |
5 |
| xyz file |
xyz file |
xyz file |
|
Figure 2. B3LYP/6-31G(d) optimized structures of 3-5. Distances (Å) shown are between C1-C6 and C3-C4 of the hexadiene component of the Cope rearrangement.2
|
Why does a stable bis-allyl analogue exist on the Cope reaction surface of 3? In the prototype Diels-Alder reaction of 1,5-hexadiene, the possible bis-allyl intermediate (i.e., two isolated allyl radicals) is about 26 kcal mol-1 higher in energy than the Cope transition state. Only with significant radical stabilization might one expect a bis-allyl intermediate to occur. One can consider 5 as composed of two bridged phenalenyl radicals (6). Phenalenyl radical is stable due to electron delocalization; its ESR spectrum has been observed, but it has not been isolated, instead dimerizing to give 7.3 In addition to the stabilization afforded by the extensive delocalization of the radical within the phenalenyl system, two phenalenyl systems can also interact through overlap of their π-systems, creating what has been termed π-dimerization.4-6 MRMP2 computations suggest that the π-dimerization energy of 6 is 11 kcal mol-1.7 While the geometry of 5 is not ideal for π-dimerization, its structure clearly indicates some stacking of the two phenalenyl fragments. Both the enhanced electron delocalization about the large phenalenyl system along with π-dimerization provides sufficient stabilization that the bis-allyl intermediate exits on the Cope rearrangement pathway. This now completes all of the options for how the Cope rearrangement may occur: either directly through a concerted transition state, or multi-step process with a 1,6-diyl intermediate or a bis-allyl intermediate.

Cope Rearrangement of 3-Vinylmethylenecyclobutane
3-Vinylmethylenecyclobutane 8 can undergo a myriad of thermal rearrangements involving [1,3]- and [3,3]-shifts.8 The Cope rearrangement of 8 to 9 has a barrier of 35.7 kcal mol-1.9 This large barrier is consistent with cleavage of a C-C bond leading to a diradical intermediate.

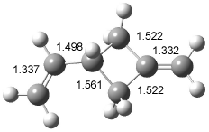
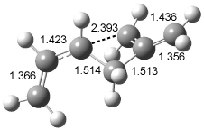
Houk has recently confirmed the diradical nature of this rearrangement.10 The geometries of all reactants intermediates, products and transition states were optimized at UB3LYP/6-31+G(d) and single-point energies were evaluated at CASPT2(6,6)/6-31G(d). Two diradical intermediates, 10 and 11, lie 30.0 and 32.0 kcal mol-1, respectively, above 8. These intermediates are separated by a small barrier, 1.5 kcal mol-1 from 10, and a barrier of 2.0 kcal mol-1 interconverts mirror versions of 10. All of these paths are sketched in Scheme 3 and the geometries of the critical points are displayed in Figure 3.
Scheme 3

Figure 3. Optimized Structures of the critical points in Scheme 3.10
Only the reaction going forward from 11 can lead to product 9. There are two such routes, involving an exo or endo approach. They are of similar energy, and also very close in energy to that of the diradical intermediates. Houk concludes that the diradical intermediates “have substantial conformational freedom and very low barriers for forming stereo- and regioisomeric forms of the ring-enlarged product”, in agreement with the experimentally observed lack of any region- or stereoselectivity in the thermal reactions of 8. The computed barrier, 34.9 kcal mol-1 for TS8-10, is in good accord with the experimental barrier of 35.7 kcal mol-1.
A study by Jung11 the year before actually inspired Houk’s work. Jung discovered that appropriately substituted vinylmethylcyclohexenes will undergo very selective Cope rearrangements; for example, thermolysis of 8a produces 9a in greater than 90% yield. This result is quite contrary to that normally observed for vinylmethylcyclohexene thermoylsis: many products with virtually complete scrambling of all stereochemical information.

Examination of the rearrangement of 8a is computationally prohibitive, so Houk looked at the effect of individual substituents. The role of the trialkylsiloxy group was evaluated through the rearrangement of 8b, leading to diradicals 10b and 11b (Scheme 4). The transition state leading to 11b is 1.5 kcal mol-1 below that leading to 10b. This is opposite the relative ordering of the transition states in the parent reaction, indicating that siloxy substitution would favor the path that leads to direct Cope rearrangement, which must pass through 11. The preference for the opposite TS with the siloxy group results from its torquoselectivity (See Chapter 3.5) Since 11b is more stable than 10b, this would also help preserve the stereochemistry during the rearrangement.
Scheme 4

The effects of the terminal substituents were also evaluated. As shown in Scheme 5, the Cope rearrangement of 8b is predicted to proceed with distinct stereoselectivity. The ring opening step preferentially produces diradical intermediate 12 over 13. The ring forming step is also stereoselective: 12 cyclizes to 14 in a 3:1 ratio, while the ring closure of 13 predominantly gives 15. Overall, the rearrangement of 8b is predicted to give a product ratio 14:15 of 2:1. This is in accord with the Jung’s experimental observation.
Scheme 5

References
(1) Navarro-Vazquez, A.; Prall, M.; Schreiner, P. R., “Cope Reaction Families: To Be or Not to Be a Biradical,” Org. Lett. 2004, 6, 2981-2984, DOI: 10.1021/ol0488340
(2) Huang, J.; Kertesz, M., “Stepwise Cope Rearrangement of Cyclo-biphenalenyl via an Unusual Multicenter Covalent π-Bonded Intermediate,” J. Am. Chem. Soc. 2006, 128, 7277-7286, DOI: 10.1021/ja060427r
(3) Zheng, S.; Lan, J.; Khan, S. I.; Rubin, Y., “Synthesis, Characterization, and Coordination Chemistry of the 2-Azaphenalenyl Radical,” J. Am. Chem. Soc. 2003, 125, 5786-5791, DOI: 10.1021/ja029236o
(4) Goto, K.; Kubo, T.; Yamamoto, K.; Nakasuji, K.; Sato, K.; Shiomi, D.; Takui, T.; Kubota, M.; Kobayashi, T.; Yakusi, K.; Ouyang, J., “A Stable Neutral Hydrocarbon Radical: Synthesis, Crystal Structure, and Physical Properties of 2,5,8-Tri-tert-butyl-phenalenyl,” J. Am. Chem. Soc. 1999, 121, 1619-1620, DOI: 10.1021/ja9836242
(5) Suzuki, S.; Morita, Y.; Fukui, K.; Sato, K.; Shiomi, D.; Takui, T.; Nakasuji, K., “Aromaticity on the Pancake-Bonded Dimer of Neutral Phenalenyl Radical as Studied by MS and NMR Spectroscopies and NICS Analysis,” J. Am. Chem. Soc. 2006, 128, 2530-2531, DOI: 10.1021/ja058387z
(6) Takano, Y.; Taniguchi, T.; Isobe, H.; Kubo, T.; Morita, Y.; Yamamoto, K.; Nakasuji, K.; Takui, T.; Yamaguchi, K., “Hybrid Density Functional Theory Studies on the Magnetic Interactions and the Weak Covalent Bonding for the Phenalenyl Radical Dimeric Pair,” J. Am. Chem. Soc. 2002, 124, 11122-11130, DOI: 10.1021/ja0177197
(7) Small, D.; Zaitsev, V.; Jung, Y.; Rosokha, S. V.; Head-Gordon, M.; Kochi, J. K., “Intermolecular π-to-π Bonding between Stacked Aromatic Dyads. Experimental and Theoretical Binding Energies and Near-IR Optical Transitions for Phenalenyl Radical/Radical versus Radical/Cation Dimerizations,” J. Am. Chem. Soc. 2004, 126, 13850-13858, DOI: 10.1021/ja046770i
(8) Kozhushkov, S. I.; Kuznetsova, T. S.; Zefirov, N. S., “Mechanism of Thermal Isomerization of 3-Vinylmethylenecyclobutane into 4-Methylenecyclohexane,” Dokl. Akad. Nauk SSSR, 1988, 299, 1395-1399,
(9) Dolbier, W. R.; Mancini, G. J., “Non-concerted Thermal Reorganizations 3,3-Divinylmethylenecyclobutane,” Tetrahedron Lett. 1975, 16, 2141-2144, DOI: 10.1016/S0040-4039(00)72661-X.
(10) Zhao, Y. L.; Suhrada, C. P.; Jung, M. E.; Houk, K. N., “Theoretical Investigation of the Stereoselective Stepwise Cope Rearrangement of a 3-Vinylmethylenecyclobutane,” J. Am. Chem. Soc. 2006, 128, 11106-11113, DOI: 10.1021/ja060913e
(11) Jung, M. E.; Nishimura, N.; Novack, A. R., “Versatile Diastereoselectivity in Formal [3,3]-Sigmatropic Shifts of Substituted 1-Alkenyl-3-alkylidenecyclobutanols and Their Silyl Ethers,” J. Am. Chem. Soc. 2005, 127, 11206-11207, DOI: 10.1021/ja051663p
InChI
3: InChI=1/C34H26/c1-19-21-11-12-22-16-24-8-6-10-26-18-28-14-13-27-17-25-9-5-7-23(15-21)29(25)31(19)33(27,3)34(28,4)32(20(22)2)30(24)26/h5-18H,1-4H3/b12-11-/t33-,34+
5: InChI=1/C34H26/c1-19-23-11-12-25-17-29-9-6-10-30-18-26(22(4)32(21(25)3)34(29)30)14-13-24-16-28-8-5-7-27(15-23)33(28)31(19)20(24)2/h5-18H,1-4H3/b12-11-,14-13-
6: InChI=1/C13H9/c1-4-10-6-2-8-12-9-3-7-11(5-1)13(10)12/h1-9H
7: InChI=1/C26H18/c1-5-17-9-3-11-23-21(15-13-19(7-1)25(17)23)22-16-14-20-8-2-6-18-10-4-12-24(22)26(18)20/h1-16,21-22H
8: InChI=1/C7H10/c1-3-7-4-6(2)5-7/h3,7H,1-2,4-5H2
9: InChI=1/C7H10/c1-7-5-3-2-4-6-7/h2-3H,1,4-6H2