I have discussed the circulenes in a few previous posts. Depending on their size, they can be bowls, flat disks, or saddles. A computational study of [7]circulene noted that C2 structure is slightly higher in energy than the Cs form,1 though the C2 form is found in the x-ray structure.2
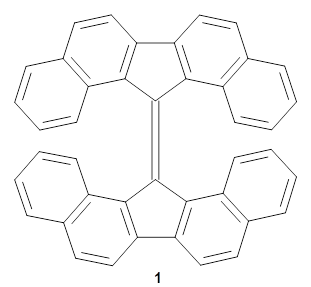
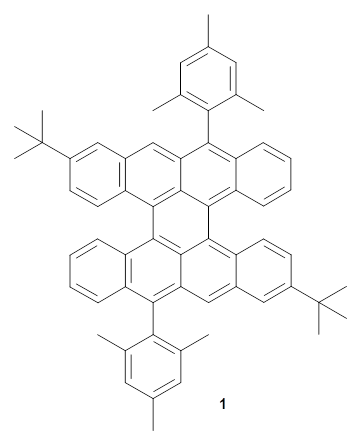
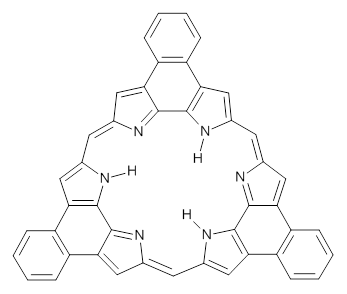
Now, Miao and co-workers have synthesized the tetrabenzo[7]circulene 1 and also examined its structure using DFT.3
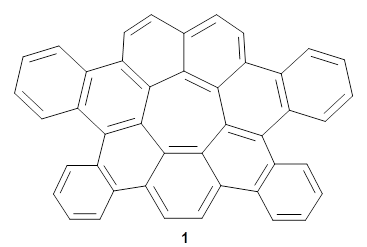
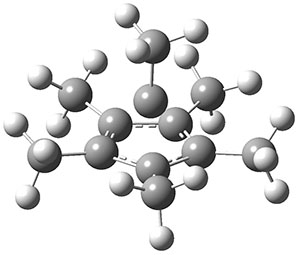
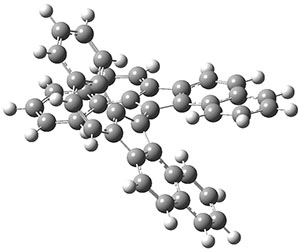
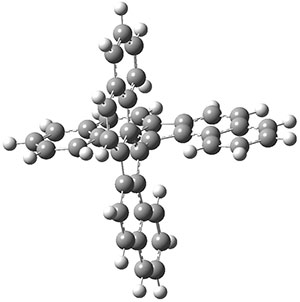
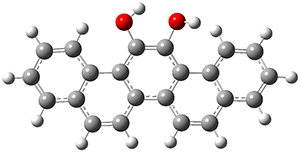
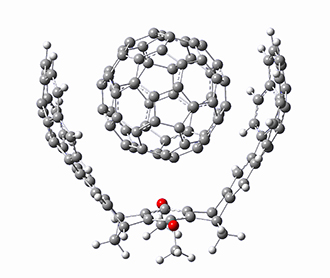
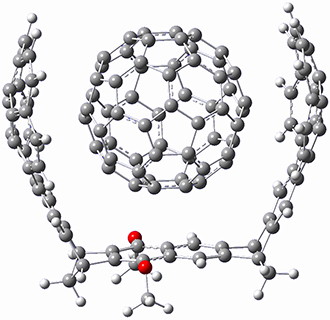
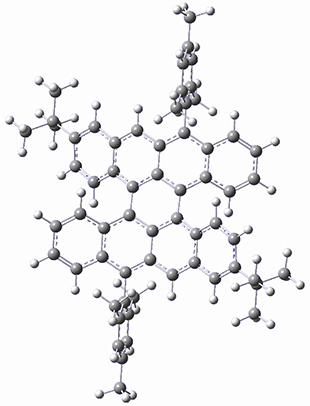
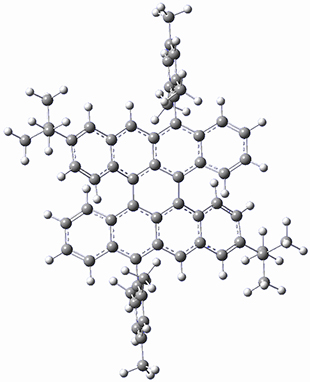
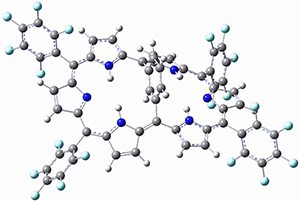
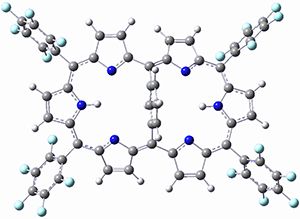
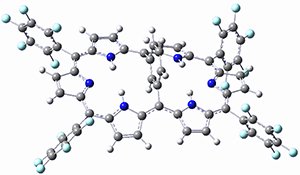
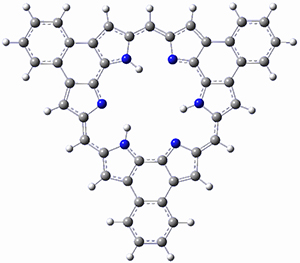
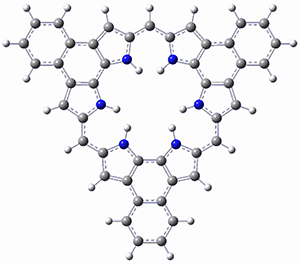
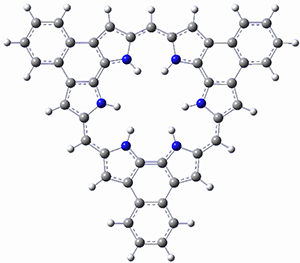
As with the parent compound, a C2 and Cs form were located at B3LYP/6-31G(d,p), and are shown in Figure 1. The C2 form is 7.6 kcal mol-1 lower in energy than the Cs structure, and the two are separated by a transition state (also shown in Figure 1) with a barrier of 12.2 kcal mol-1. The interconversion of these conformations takes place without going through a planar form. The x-ray structure contains only the C2 structure. It should be noted that the C2 structure is chiral, and racemization would take place by the path: 1-Cs ⇆ 1-Cs ⇆ 1-C2*, where 1-C2* is the enantiomer of 1-C2.
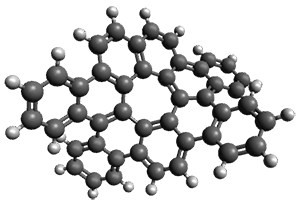
1-C2 |
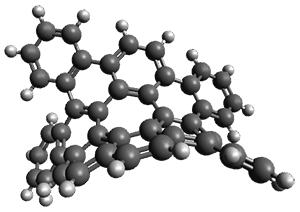
1-TS |
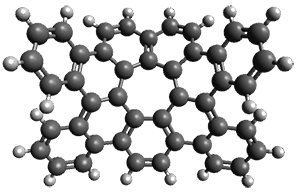
1-Cs |
Figure 1. B3LYP/6-31G(d,p) optimized structures of 1.
References
1) Hatanaka, M., "Puckering Energetics and Optical Activities of [7]Circulene Conformers." J. Phys. Chem. A 2016, 120 (7), 1074-1083, DOI: 10.1021/acs.jpca.5b10543.
2) Yamamoto, K.; Harada, T.; Okamoto, Y.; Chikamatsu, H.; Nakazaki, M.; Kai, Y.; Nakao, T.; Tanaka, M.; Harada, S.; Kasai, N., "Synthesis and molecular structure of [7]circulene." J. Am. Chem. Soc. 1988, 110 (11), 3578-3584, DOI: 10.1021/ja00219a036.
3) Gu, X.; Li, H.; Shan, B.; Liu, Z.; Miao, Q., "Synthesis, Structure, and Properties of Tetrabenzo[7]circulene." Org. Letters 2017, DOI: 10.1021/acs.orglett.7b00714.
InChIs
1: InChI=1S/C44H22/c1-5-13-28-24(9-1)32-19-17-23-18-20-33-25-10-2-6-14-29(25)38-31-16-8-4-12-27(31)35-22-21-34-26-11-3-7-15-30(26)37(28)43-39(32)36(23)40(33)44(38)42(35)41(34)43/h1-22H
InChIKey=KVMXYGAVHDZMNP-UHFFFAOYSA-N