The conformational preference of α-β-unsaturated carbonyl compounds is well established: the two π-bonds prefer to be in conjugation with the oxygen and three carbon atoms (nearly) coplanar. Now, what about the conformational preference of vinyl sulfoxides? Since the S-O π-bond is weak, alternate conformations might be favorable. Podlech has prepared some 1,3-dithian-1-oxides that should be conformationally static and thereby offer some insight into this question.1 The dithiane oxides 1 and 2 can exist with the S-O bond in the axial (a) or equatorial (e) positions.
|
|
|


The B3LYP/6-31++G(d,p) geometries are shown in Figure 1. The equatorial structure has the two π bonds close to coplanar (the C-C-S-O dihedral is 14°), while in the axial isomers, the C-C-S-O dihedral is about -122°.
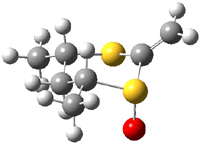 1a |
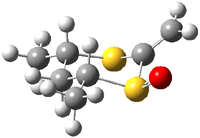 1e |
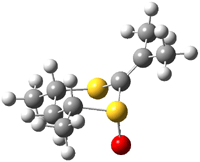 2a |
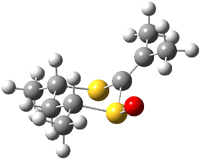 2e |
Figure 1. B3LYP/6-31++G(d,p) optimized structures of 1 and 2.
Podlech argues for a πC=C → σ*S-O stabilization in the axial isomer on the basis of two observations. First, the UV maximum absorbance in 1a is at 266nm, 12 nm greater than in 1e and similarly, the UV maximum in 2a is 2 nm higher than in 2e. Second, NBO analysis indicates a much larger contribution for this interaction in 1a (3.05 kcal mol-1) than in 1e (0.07 kcal mol-1).
However, I am unconvinced that this interaction is really dominant. Oxidation of the precursor dithiane with MCPBA gives a 42:58 ratio of 1e:1a and a 76:24 ratio of 2e:2a, which indicates a preference for the equatorial form of 1 and only a small preference for the axial form of 2. Unreported by Podlech (even in the supporting materials) is the relative computed energy difference of the two stereoisomers. At B3LYP/6-31++G(d,p) with ZPE, 1e is 2.6 kcal mol-1 lower in energy than 1a and 2e is 0.05 kcal mol-1 lower than 2a. So, in the gas-phase, it appears that the vinyl sulfoxides prefer the equatorial orientation, just as in α-β-unsaturated carbonyl compounds. The πC=C → σ*S-O interaction is stronger in the axial conformation, but it is doubtful that this alone manifests in any diastereomeric selectivity.
References
(1) Ulshöfer, R.; Podlech, J., "Stereoelectronic Effects in Vinyl Sulfoxides," J. Am. Chem. Soc. 2009, 131, 16618-16619, DOI: 10.1021/ja904354g

Eutactic responded on 26 Jan 2010 at 2:26 am #
Can’t read the paper from here, but the πC=C → σ*S-O interaction is very interesting indeed. It would be interesting to see how PCM changes the picture (or not) with respect to the charge distribution on the S=O bond…
Also near the end should it read “2e is 0.05 kcal mol-1 lower than 2a”?
Steven Bachrach responded on 26 Jan 2010 at 6:15 am #
Corrected. Thanks!
Henry Rzepa responded on 28 Jan 2010 at 5:17 am #
The π-CC → σ*S-O interaction may have other analogues. Thus in this post a possible π-CC → σ*C-CN interaction was mooted, along with a much stronger lp(O) → σ*C-CN anomeric effect. In the post, it was observed that the possibility of such interactions was not commented on in the original article; I suspect many such are missed, when the focus is purely on the synthetic aspects of a molecule.