Herges and co-workers have prepared a triply-twisted Möbius molecule. 1 The key element is recognizing that most of the “twisting” needs to be accomplished through writhe, a twisting that produces figure-8-like crossing, the way an old-school phone cord twists about itself or the way a pretzel is formed. Herges employs three bi-naphthelene subunits to provide the template for the writhe needed. The prepared compound is 1. A clever, relatively straightforword synthesis produces this amazing molecule, along with the single-twisted 2.
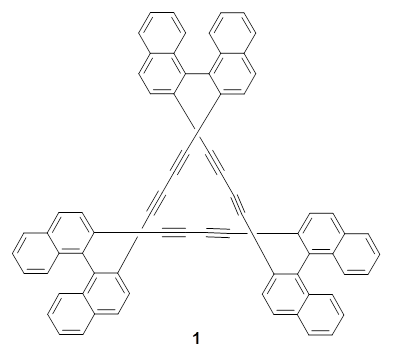
The B3LYP/6-31G* optimized geometries of 1 and the single-twisted analogue 2 are shown in Figure 1. Table 1 presents the key topological parameters of 1 and 2, comparing the computed and X-ray crystal structure. The absolute value of the linking number Lk is 3, indicating the three twists, and the reason that this highly twisted molecule can be made is that half of the twist actually results from writhe.

1 |

2 |
Figure 1. B3LYP/6-31G* optimized geometries of the two diastereomers if 1. (Be sure to click on these images to launch JMol and interactively manipulate the structures!)
Table 1. Topological parameters of 1. a
|
|
|Lk| |
|Tw| |
|Wr| |
|
X-ray, 1 |
3.0 |
1.42 |
1.58 |
|
Comp, 1 |
3.0 |
1.33 |
1.67 |
|
Comp, 2 |
1.0 |
0.37 |
0.63 |
aLk is the linking number, Tw is the twist number, and Wr is the writhe number, with the condition that Tk + Wr = Lk.
References
(1) Schaller, G. R.; Topić, F.; Rissanen, K.; Okamoto, Y.; Shen, J.; Herges, R. "Design and synthesis of the first triply twisted Möbius annulene," Nat. Chem. 2014, 6, 608-613, DOI: 10.1038/nchem.1955.
InChIs
1: InChI=1S/C72H36/c1-2-26-56-44-38-50-20-8-14-32-62(50)68(56)70-58(46-40-52-22-10-16-34-64(52)70)28-5-6-30-60-48-42-54-24-12-18-36-66(54)72(60)71-59(47-41-53-23-11-17-35-65(53)71)29-4-3-27-57-45-39-51-21-9-15-33-63(51)69(57)67-55(25-1)43-37-49-19-7-13-31-61(49)67/h7-24,31-48H/b69-67-,70-68-,72-71-
InChIkey=ICOGWBLQQNBWJA-UWFYZODKSA-N

Henry Rzepa responded on 13 Oct 2014 at 1:01 pm #
The idea that destabilising twist can be converted to writhe is six years old now (see for example DOI: 10.1021/ja710438j). Many known hexaphyrins have more or less equal distributions of twist and writhe (see DOI: 10.1021/ol703129z from 2008) and in 2010 I proposed a decapentaphyrin which amazingly has more than all its twist converted to writhe. Thus this molecule is predicted to have Lk =6π, Tw = ~-0.8π and Wr ~ +6.8π (yes the two can be either positive or negative, and do not have to be the same sign).
Of course there is a difference between proposing and making a molecule. But can I suggest the decapentaphyrin alluded to here as a synthetic challenge?
Henry Rzepa responded on 13 Oct 2014 at 1:07 pm #
Steve, I think you have an error in the line
Comp, 2 3.0 0.37 0.63
The 3 refers to Lk. For a single twisted isomer, it should be 1.0, not 3.0.
Henry Rzepa responded on 13 Oct 2014 at 1:30 pm #
On more query. You show |Lk| |Tw| |Wr| i.e. the modulus of each topological invariant. But they are in fact sign sensitive, as my example in my previously submitted comment on the decapentaphyrin shows. That is, the sign matters! Think of Lk as a member of the Cahn-Ingold-Relog conventions for chiral molecules (M and P for helical systems). Lk if you like is an extension to the simple M/P notation, in that it describes higher order helices, and -Lk and +Lk are the equivalent of M and P. But since Lk can be decomposed into Tw and Wr, the signs there also matter. Taking the modulus would destroy vital information!
Steven Bachrach responded on 13 Oct 2014 at 1:42 pm #
Thanks Henry for catching the error for the Lk value for 2. That has now been corrected.
Also, Henry is correct that this paper is not the first to propose using writhe as a means for creating highly twisted compounds. Nonetheless, this paper does offer the preparation of a highly twisted species made possible through significant writhe, confirmation of previous proposals.
Henry Rzepa responded on 13 Oct 2014 at 3:54 pm #
One of the reasons for providing coordinates of molecules the way Steve does is that they can be inspected much more readily (the alternative is to extract the information from e.g. a PDF file in supporting information). If one does precisely this for structure 1, and measures the dihedral angle across the dinaphthyl C-C bond, it emerges at around 79°. The length is 1.50Å. Neither value is commensurate with the description aromatic molecule as e.g. used in the title of this post, but instead indicates a close orthogonality of the π-system at any of these three bonds. Surely 1.5Å indicates an interruption of conjugation, and hence of any putative aromaticity? An aromatic value of course would be closer to 1.4Å.
Steve, is it possible in your diagrams to annotate structures with such values? Thus a script of the form measure 1 2; will in fact throw up the relevant bond length in the 3D model that appears when you click on your diagrams?
Regarding my comment about |Lk| |Tw| |Wr|, perhaps I can clarify it. One should write |Lk= Tw + Wr|, that is to say the modulus should apply to the whole expression, not the individual components, and it is the value of Lk that is ± (to indicate individual enantiomers of the D3-disymmetric molecule).
Henry Rzepa responded on 14 Oct 2014 at 4:50 am #
More thoughts about the topology of molecule 1. In calculating a linking number, one sums the local twists about each bond in the annulene, and then adds the writhe to get the final topology. If any local twist is 90° one however has a mathematical singularity. Normally, the twists are all in one direction (ending with a chiral object), but at 90° the direction of twist is in effect equally probable in either a left or right handed sense. So for molecules which contain local twists close to 90°, the topology becomes in effect unstable/undeterminable. Molecule 1 has three local twists of 79° which is therefore close to becoming topologically unstable.
The ideal Möbius system would therefore not be topologically unstable. For a 24π-electron system, such an ideal system should be fully delocalised with C-C bond lengths close to 1.4Å (for odd values of Lk, these follow a 4n rule, and even values of Lk follow a 4n+2 rule, see for example DOI: 10.1016/j.comptc.2014.09.028). Of course, in the limit of a very large annulene, just as with poly-acetylene, the bonds will start to alternate. This effect is thought to start at around 26 π-electrons for flat systems (Lk = 0), but there are hints that it might start much later for systems with a linking number Lk ≠ 0. This being just one reason why further syntheses/discoveries of such topologically twisted molecules are very important.
The holy grail now must be to find a topologically stable example to add to molecule 1. The decapentaphyrin noted earlier is certainly topologically stable.