In section 4.4 of the book, I discuss in great detail the computational (and some experimental) studies of the benzynes, the formal diradicals created by loss of two hydrogen atoms from benzene. Now comes a very nice experimental study on a molecule that takes the next step: 1,3,5-tridehydrobenzene 1, benzene that lacks three hydrogen atoms. Sander reports the preparation and characterization of trifluoro-1,3,5-tridehydrobenzene 2.1 The characterization of this novel molecule is made through comparison with computed IR spectra.
2 is prepared by flash vapor pyrolysis of 1,3,5-triiodo-2,4,6-trifluorobenezene
and then trapping the products in a low temperature matrix. Sander identifies five IR peaks of a product he believes is 2. These IR frequencies are listed in Table 1.
Table 1. Experimental and computeda IR frequencies (cm-1) and relative intensities of 2.
|
Expt |
2a |
2bb |
|||
|
ν |
I |
ν |
I |
ν |
I |
|
954 |
60 |
921.7 |
57 |
976.2 |
57 |
|
1030 |
30 |
997.6 |
54 |
1016.0 |
55 |
|
1266 |
40 |
1221.8 |
35 |
1291.3 |
33 |
|
|
|
1310.6 |
16 |
1325.4 |
30 |
|
1560 |
70 |
1530.0 |
73 |
1572.6 |
100 |
|
1738 |
100 |
1726.6 |
100 |
1690.6 |
88 |
aUBLYP/cc-pVTZ. bTransition state.
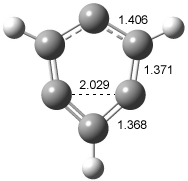
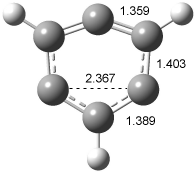
In order to confirm that this IR spectra comes from 2, Sander computed the structure and IR frequencies of both 1 and 2. The 2A1 structure of 1 had been studied previously2, but what had gone unnoticed is that another structure is possible, the 2B2 state. These two states differ in the separation between C1 and C3. When the distance is short, the SOMO is of a1 symmetry and this orbital has bonding character between these two carbon centers, giving rise to the 2A1 state (1a). As the distance gets longer between C1 and C3, a b2 orbital, having antibonding character between C1and C3, becomes lower in energy than the a1 orbital, so that the structure is 2B2 (1b). The UBLYP/cc-pVTZ optimized structures are shown in Figure 1. 1a is 2-3 kcal mol-1 lower in energy than 1b. Furthermore, 1b has one imaginary frequency and is not a local energy minimum. Sander also optimized the structures of 2a and 2b¸ finding little effect due to the fluorine substitution.
 1a |
 1b |
Figure 1. UBLYP/cc-pVTZ optimized structures of 1a (2A1) and 1b (2B1).
The computed IR frequencies are listed in Table 1. The computed frequencies (and their relative intensities) of 2a match up strikingly well with those of the experiment. Sander concludes that 2a has in fact been prepared and characterized.
References
(1) Venkataramani, S.; Winkler, M.; Sander, W., "Trifluoro-1,3,5-tridehydrobenzene," Angew. Chem. Int. Ed. 2007, 46, 4888-4893, DOI: 10.1002/anie.200700536
(2) Cristian, A. M. C.; Shao, Y.; Krylov, A. I., "Bonding Patterns in Benzene Triradicals from Structural, Spectroscopic, and Thermochemical Perspectives," J. Phys. Chem. A 2004, 108, 6581-6588, DOI: 10.1021/jp049007j.
InChI:
1: InChI=1/C6H3/c1-2-4-6-5-3-1/h1,4-5H
2: InChI=1/C6F3/c7-4-1-5(8)3-6(9)2-4
